23 gesichtete, geschützte Fragmente: Plagiat
| [1.] Hw/Fragment 002 01 - Diskussion Bearbeitet: 31. August 2014, 14:07 Singulus Erstellt: 27. August 2014, 20:21 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 2, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 13, 14, Zeilen: 13: 20ff; 14:1ff |
|---|---|
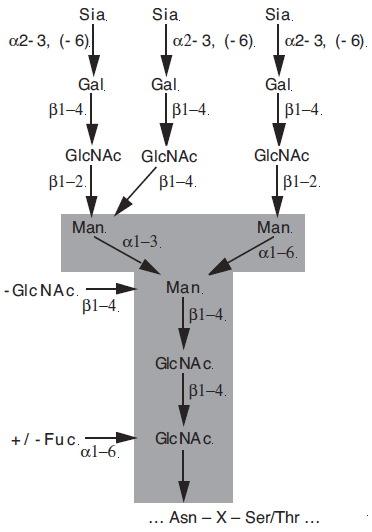
| N-Glycane besitzen eine gemeinsame Core-Struktur aus zwei N-Acetylglucosaminen (GlcNAc) und drei Mannoseresten. Über einen N-Acetylglucosaminrest wird diese Core-Struktur an Asparagin in der Konsensussequenz –Asn-Xxx-Ser/Thr von Glycoproteinen gebunden. Es lassen sich drei Arten des variablen Strukturteils unterscheiden: mannosereiche N-Glycane besitzen neben der Core-Struktur nur noch Mannosereste; die des komplexen Typs besitzen zusätzlich N-Acetyllactosamineinheiten, Fucosen und Sialinsäuren; der hybride Typ stellt eine Mischform des mannosereichen und des komplexen Typs dar. Die Strukturen der O-Glycane sind über einen N-Acetylgalactosaminrest an Serin oder Threonin von Glycoproteinen gebunden.
Abbildung 2: Grundstruktur eines typischen triantennären, komplexen N-Glycans. Die für alle N-Glycane gemeinsame Kernstruktur (GlcNAc2Man3) ist grau unterlegt. ...Asn-X-Ser/Thr... ist das Aminosäure-Sequenzmotiv für die N-Glycosylierung. Neben dem in dieser Abbildung gezeigten triantennären Glycan sind bei Kohlenhydratstrukturen des komplexen Typs zusätzlich mono- und biantennäre Formen möglich. |
N-Glycane sind über ein N-Acetylglucosamin (GlcNAc) an Asparagin in der Konsensussequenz Asn-X-Ser/Thr von Glycoproteinen gebunden und besitzen eine gemeinsame Kernstruktur aus zwei GlcNAc- und drei Mannoseresten (Abb. 1.2). Der variable Strukturteil läßt sich in drei Klassen unterteilen (Schachter, 2000). Mannosereiche N-Glycane besitzen neben der Kernstruktur nur noch Mannosereste. Die Oligosaccharide des komplexen Typs besitzen zusätzlich N-Acetyllactosamineinheiten, Fucosen und Sialinsäuren. Der hybride Typ stellt eine Mischform aus mannosereichem und komplexem Typ dar. [...]
[Seite 14]
Abbildung 1.2: Grundstruktur eines typischen triantennären, komplexen N-Glycans. Die für alle N-Glycane gemeinsame Kernstruktur (GlcNAc2Man3) ist grau unterlegt. ...Asn-X-Ser/Thr... ist das Aminosäure-Sequenzmotiv für die N-Glycosylierung. Neben dem in dieser Abbildung gezeigten triantennären Glycan sind bei Kohlenhydratstrukturen des komplexen Typs zusätzlich mono- und biantennäre Formen sowie auch tetra- und pentaantennäre Formen möglich (Fukuda, 1994). O-Glycane sind über N-Acetylgalactosamin (GalNAc) an einen Serin- oder Threoninrest von Glycoproteinen gebunden. |

Ein Verweis auf die Quelle fehlt. |
|
| [2.] Hw/Fragment 006 15 - Diskussion Bearbeitet: 31. August 2014, 14:00 Singulus Erstellt: 27. August 2014, 20:11 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 6, Zeilen: 15-25 |
Quelle: Blume 2003 Seite(n): 16, Zeilen: 5ff |
|---|---|
| Die größte Familie Sialinsäure-bindender Lektine in Säugetieren bilden die Siglecs. Beim Menschen sind 13 verschiedene Siglecs gefunden worden. Die meisten befinden sich auf den Zellen des Immunsystems. Näher untersucht wurde zum Beispiel Siglec1/Sialoadhäsin, welches ausschließlich von Makrophagen exprimiert wird und die Interaktion dieser Zellen mit anderen Zellen des Immunsystems über die Bindung α2,3-gebundener Sialinsäuren reguliert (Hartner et al., 2001). Siglec2/CD22 ist an der homophilen Interaktionen von B-Zellen beteiligt und bindet ausschließlich α2,6-verknüpfte Sialinsäuren (Tedder et al., 1997).
Andererseits nutzen auch Pathogene Sialinsäuren als Bindungspartner auf ihren Zielzellen. So sind virale Hämagglutinine sialinsäurebindende Lektine, welche die Agglutination von Erythrozyten vermitteln können. Das Hämagglutinin des Influenza A-Virus ist das derzeit am besten untersuchte. Seine Spezifität für bestimmte Sialinsäuretypen ist streng abhängig von der [Sialylierung der Wirtszelle, z.B. von Mensch, Huhn oder Schwein (Suzuki et al., 2000).] |
Beim Menschen sind 11 verschiedene Siglecs gefunden worden, die meisten finden sich auf den Zellen des Immunsystems. Die Funktionen der einzelnen Siglecs sind bis heute nur unzureichend geklärt; einige wenige, seit längerem bekannte Vertreter sind jedoch intensiver untersucht worden. Siglec1/Sialoadhäsin wird ausschließlich auf Makrophagen exprimiert und reguliert die Interaktion dieser Zellen mit anderen Zellen des Immunsystems über die Bindung α2,3-gebundener Sialinsäuren (Hartnell et al., 2001). Siglec2/CD22 ist an der homophilen Interaktion von B-Zellen beteiligt und bindet ausschließlich α2,6-gebundene Sialinsäuren (Tedder et al., 1997). [...]
[...] Nicht nur endogene sialinsäurebindende Lektine eines multizellulären Organismus nutzen Sialinsäuren als Bindungspartner auf ihren Zielzellen, sondern auch Pathogene, wie Viren, Bakterien und Parasiten. So besitzen Viren Hämagglutinine, sialinsäurebindende Lektine, die die Agglutination von Erythrozyten (Hämozyten) vermitteln können. Das am besten untersuchte Hämagglutinin ist das des Influenza A-Virus. Seine Spezifität für bestimmte Sialinsäuretypen hängt streng von der Sialylierung der Wirtszelle (z.B. von Mensch, Huhn oder Schwein) ab (Suzuki et al., 2000). |
Ein Verweis auf die Quelle fehlt. Fortsetzung auf der folgenden Seite: Hw/Fragment 007 01 |
|
| [3.] Hw/Fragment 040 01 - Diskussion Bearbeitet: 31. August 2014, 12:58 Schumann Erstellt: 27. August 2014, 09:03 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 40, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 40, Zeilen: 44: 20ff; 45: 1ff |
|---|---|
| 6.2.2.2. Transformation von Plasmid-DNA in S. cerevisiae
Als Transformation wird die Aufnahme von Fremd-DNA und somit die genetische Veränderung von Organismen bezeichnet. Die S. cerevisiae-Zellen werden mittels der von M. Pein modifizierten Gefriermethode von R.J. Dohmen et. al. (1991) transformiert. Dafür werden die gefrorenen kompetenten S. cerevisiae-Zellen mit 2,5 μg Plasmid-DNA versetzt. Alle Ansätze werden 5 min bei 37 °C geschüttelt, mit 1,2 ml 40% (v/v) PEG 3350 (Sigma) / 200 mM Tricin- NaOH, pH 8,4, gemischt und ohne weiteres Schütteln 1 h bei 30 °C inkubiert. Nach Sedimentation bei 13000 x g für 5 sec werden die Zellen zweimal mit je 1 ml 0,15 M NaCl / 10 mM Tricin-NaOH, pH 8,4, gewaschen, anschließend in 200 μl 0,15 M NaCl/10 mM Tricin- NaOH, pH 8,4, aufgenommen, auf Agar-Platten mit Minimalmedium ausplattiert und etwa 4 Tage bis zum Erscheinen von Kolonien bei 30 °C inkubiert. Von den erhaltenen Transformanten werden einzelne Klone als Strichkolonien auf Agar-Platten mit Minimalmedium subkultiviert und für die beschriebenen Experimente eingesetzt. 6.2.2.3. Ermittlung der optimalen Expressionsbedingungen Um die optimalen Bedingungen für eine maximale Expression von rekombinantem Protein zu ermitteln, werden zunächst Pilotexpressionen durchgeführt. Einzelne Klone werden in 5 ml Minimalmedium bis zur Sättigung bei 30 °C kultiviert. Mit 500 μl dieser Vorkultur werden 50 ml Minimalmedium beimpft und bis zu einer OD600 von 1 bei 30 °C kultiviert. Durch Zugabe von 2% (w/v) Galactose wird die Proteinexpression induziert. Um die optimalen Bedingungen für maximale Expression von rekombinantem Protein zu ermitteln, werden zu verschiedenen Zeitpunkten jeweils 5 ml Aliquots abgenommen, die OD600 ermittelt und die Hefezellen durch Zentrifugation für 5 min bei 3000 x g und 4 °C pelletiert. Der Medienüberstand und die Zellen werden bis zur weiteren Analyse bei -70 °C gelagert, wobei die Zellen zuvor noch mit 500 μl Wasser gewaschen werden. Die gefrorenen Zellen werden in Lysepuffer (10 mM Natriumphosphat, pH 7,5 / 1 mM EDTA / 1 mM DTT / 1 mM PMSF) resuspendiert, so daß eine berechnete OD600 von 50-100 eingestellt wird. Anschließend wird ein gleiches Volumen an „acid-washed 0,5 mm glass beads“ (Sigma, Deutschland) zugegeben. Die Zellsuspension wird für 30 sec gevortext und anschließend 30 sec auf Eis inkubiert. Dieser Vorgang wird siebenmal wiederholt, damit die Zellen komplett lysiert sind. Zelltrümmer und unlösliche Komponenten [werden im Anschluß bei 20000 x g und RT für 10 min abzentrifugiert.] |
2.2.3.2 Transformation von Plasmid-DNA in Saccharomyces cerevisiae
Als Transformation wird die Aufnahme von Fremd-DNA und somit die genetische Veränderung von Organismen bezeichnet. Die S. cerevisiae-Hefezellen werden mittels der von M. von Pein modifizierten Gefriermethode von R.J. Dohmen et. al. (1991) transformiert. Dafür werden die gefrorenen kompetenten S. cerevisiae-Hefezellen mit 2,5 μg Plasmid-DNA versetzt. Alle Ansätze werden 5 min bei 37 °C geschüttelt, mit 1,2 ml 40% (v/v) PEG 3350 (Sigma) / 200 mM Tricine-NaOH, pH 8,4 gemischt und ohne weiteres Schütteln 1 h bei 30 °C inkubiert. Nach Sedimentation bei 13000 x g für 5 sec werden die Zellen zweimal mit je 1 ml 0,15 M NaCl / 10 mM Tricine-NaOH, pH 8,4 gewaschen, anschließend in 200 μl 0,15 M NaCl / 10 mM Tricine-NaOH, pH 8,4 aufgenommen, auf Agar-Platten mit Minimalmedium ausplattiert und etwa 4 Tage bis zum Erscheinen von Kolonien bei 30 °C inkubiert. Von den erhaltenen Transformanten werden einzelne Klone als Strichkolonien auf Agar-Platten mit Minimalmedium subkultiviert und für die beschriebenen Experimente eingesetzt. [Seite 45] 2.2.3.3 Ermittlung der optimalen Expressionsbedingungen Um die optimalen Bedingungen für eine maximale Expression von rekombinantem Protein zu ermitteln, werden zunächst Pilotexpressionen durchgeführt. Einzelne Klone werden in 5 ml Minimalmedium bis zur Sättigung bei 30 °C kultiviert. Mit 500 μl dieser Vorkultur werden 50 ml Minimalmedium beimpft und bis zu einer OD600 von 1 bei 30 °C kultiviert. Durch Zugabe von 2% (w/v) Galactose wird die Proteinexpression induziert. Um die optimalen Bedingungen für maximale Expression von rekombinantem Protein zu ermitteln, werden zu verschiedenen Zeitpunkten jeweils 5 ml Aliquots abgenommen, die OD600 ermittelt und die Hefezellen durch Zentrifugation für 5 min bei 3000 x g und 4 °C pelletiert. Der Medienüberstand und die Zellen werden bis zur weiteren Analyse bei -70 °C gelagert, wobei die Zellen zuvor noch mit 500 μl Wasser gewaschen werden. Die gefrorenen Zellen werden in Lysepuffer (10 mM Natriumphosphat, pH 7,5 / 1 mM EDTA / 1 mM DTT / 1 mM PMSF) resuspendiert, so daß eine berechnete OD600 von 50-100 eingestellt wird. Anschließend wird ein gleiches Volumen an „acid-washed 0,5 mm glass beads“ (Sigma, Deutschland) zugegeben. Die Zellsuspension wird für 30 sec gevortext und anschließend 30 sec auf Eis inkubiert. Dieser Vorgang wird siebenmal wiederholt, damit die Zellen komplett lysiert sind. Zelltrümmer und unlösliche Komponenten werden im Anschluß bei 20000 x g und RT für 10 min abzentrifugiert. |
Ein Verweis auf die Quelle fehlt. |
|
| [4.] Hw/Fragment 043 01 - Diskussion Bearbeitet: 31. August 2014, 12:50 Schumann Erstellt: 27. August 2014, 08:45 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 43, Zeilen: 1-12 |
Quelle: Blume 2003 Seite(n): 58, Zeilen: 16-29 |
|---|---|
| [Radioaktiver UDP-GlcNAc-2-]Epimerase-Assay: Beim radioaktiven Assay werden zusätzlich zu den Komponenten des nichtradioaktiven UDP-GlcNAc-2-Epimerase- Assays 1 12,5 nCi UDP-[14C]-GlcNAc in den Enzymreaktionsansatz gegeben. In der Regel wird die Enzymreaktion für 30 min bei 37 °C inkubiert, anschließend werden 350 μl Ethanol zugegeben, wodurch die Proteine denaturiert werden, d.h. die Reaktion gestoppt wird. Das entstandene 14C-ManNAc wird durch absteigende Papierchromatographie von UDP-[14C]-GlcNAc abgetrennt. Die Proben werden auf Whatman 3MMChr-Chromatographiestreifen (Herolab, Deutschland) von 2 x 48 cm aufgetragen. Die Chromatographie wird für 16-20 h mit 70% 1-Propanol / 100 mM Natriumacetat, pH 5,0, durchgeführt. Die getrockneten Chromatographiestreifen werden anschließend in 2,5 cm lange Stücke geschnitten und die Radioaktivität mit 5 ml Ultima Gold XR (Packard, Niederlande) in einem Tri-Carb 1900 CA Flüssigszintillationszähler (Packard, Niederlande) bestimmt. Der Rf- Wert für UDP-GlcNAc und ManNAc beträgt unter diesen Bedingungen 0,08 bzw. 0,55. | Radioaktiver UDP-GlcNAc-2-Epimerase-Assay: Beim radioaktiven Assay werden zusätzlich zu den Komponenten des nichtradioaktiven UDP-GlcNAc-2-Epimerase- Assays 1 12,5 nCi UDP-[14C]-GlcNAc in den Enzymreaktionsansatz gegeben. In der Regel wird die Enzymreaktion für 30 min bei 37 °C inkubiert, anschließend werden 350 μl Ethanol zugegeben, wodurch die Proteine denaturiert werden, d.h. die Reaktion gestoppt wird. Das entstandene 14C-ManNAc wird durch absteigende Papierchromatographie von UDP-[14C]-GlcNAc abgetrennt. Die Proben werden auf Whatman 3MMChr-Chromatographiestreifen (Herolab, Deutschland) von 2 x 48 cm aufgetragen. Die Chromatographie wird für 16-20 h mit 70% (v/v) 1-Propanol / 100 mM Natriumacetat, pH 5,0 durchgeführt. Die getrockneten Chromatographiestreifen werden anschließend in 2,5 cm lange Stücke geschnitten und die Radioaktivität mit 5 ml Ultima Gold XR (Packard, Niederlande) in einem Tri-Carb 1900 CA Flüssigszintillationszähler (Packard, Niederlande) bestimmt. Der Rf-Wert für UDP-GlcNAc und ManNAc beträgt unter diesen Bedingungen 0,08 bzw. 0,55. |
Ein Verweis auf die Quelle fehlt. |
|
| [5.] Hw/Fragment 042 01 - Diskussion Bearbeitet: 31. August 2014, 12:49 Schumann Erstellt: 27. August 2014, 08:52 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 42, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 54, 57, 58, Zeilen: 54: 1ff; 57: 24ff; 58: 1ff |
|---|---|
| 6.2.3. Allgemeine proteinbiochemische Methoden
6.2.3.1. Proteinbestimmung nach Bradford Die Proteinbestimmung wird nach der Methode von Bradford (1976) durchgeführt, bei der der Farbstoff Coomassie-Brillantblau mit Proteinen unter Komplexbildung reagiert. 20 μl proteinhaltige Probe werden mit 1 ml Bradford-Reagenz (10% (v/v) Phosphorsäure / 5% (v/v) Ethanol / 0,1% (w/v) Coomassie G-250) versetzt und 3 min bei RT inkubiert. Anschließend wird die Extinktion bei 578 nm bestimmt. Als Proteinstandard dient Rinderserumalbumin. 6.2.3.2. UDP-GlcNAc-2-Epimerase-Assays Zum Nachweis der UDP-GlcNAc-2-Epimerase-Aktivität werden zum einen der nichtradioaktive UDP-GlcNAc-2-Epimerase-Assay mit anschließendem colorimetrischen Nachweis des entstandenen ManNAc zum anderen der sehr viel empfindlichere radioaktive UDP-GlcNAc-2- Epimerase-Assay durchgeführt. Nichtradioaktiver UDP-GlcNAc-2-Epimerase-Assay 1: 100 μl Probe/Wasser werden mit 45 μl 200 mM Natriumphosphatpuffer, pH 7,5, 45 μl 50 mM MgCl2 und 2,5 μl 100 mM UDP-GlcNAc gemischt und in der Regel für 30 min bei 37 °C inkubiert. Die Reaktion wird durch 1-minütige Inkubation bei 100 °C gestoppt. Die denaturierten Proteine aus dem Enzymreaktionsansatz werden durch Zentrifugation bei 20000 x g für 1 min pelletiert. Das entstandene ManNAc wird mittels Morgan-Elson-Test (Reissig et al., 1955), mit dem spezifisch N-Acetylhexosamine detektiert werden, nachgewiesen. Nach der Zentrifugation werden 150 μl des Überstandes mit 30 μl 0,8 M H2BO3-Puffer, pH 9,1 (mit KOH eingestellt), gemischt und für 3 min bei 100 °C inkubiert. Anschließend werden 800 μl Farbreagenz (1% (w/v) 4- Dimethylaminobenzaldehyd / 1,25% (v/v) 10 M HCl in Essigsäure) zugegeben und für 30 min bei 37 °C inkubiert. Die Extinktion wird bei 578 nm gemessen. Nichtradioaktiver UDP-GlcNAc- 2-Epimerase-Assay 2: Für einige zeitabhängige Aktivitätsmessungen wird ein modifizierter UDP-GlcNAc-2-Epimerase-Assay verwendet. In diesem Epimerase-Assay 2 werden unterschiedliche Probenvolumina mit 260 μl 200 mM Tris-HCl, pH 8,1 / 65 mM MgCl, 10 μl 100 mM UDP-GlcNAc, 40 μl frisch angesetztem 100 mM Phosphoenolpyruvat, 20 μl frisch angesetztem 15 mM NADH und 2 μl Pyruvat-Kinase/Lactat-Dehydrogenase (2 U) in einem Gesamtvolumen von 800 μl gemischt. Die Enzymreaktion wird bei 37 °C im Photometer bei 340 nm verfolgt. Als Kontrolle wird ein Ansatz ohne Enzym gemessen und die ermittelten Werte später von den Werten der Enzymreaktion abgezogen. |
2.2.6 Allgemeine proteinbiochemische Methoden
2.2.6.1 Proteinbestimmung nach Bradford Die Proteinbestimmung wird nach der Methode von Bradford (1976) durchgeführt, bei der der Farbstoff Coomassie-Brillantblau mit Proteinen unter Komplexbildung reagiert. 20 μl proteinhaltige Probe werden mit 1 ml Bradford-Reagenz (10% (v/v) Phosphorsäure / 5% (v/v) Ethanol / 0,1% (w/v) Coomassie G-250) versetzt und 3 min bei RT inkubiert. Anschließend wird die Extinktion bei 578 nm bestimmt. Als Proteinstandard dient Rinderserumalbumin. [Seite 57] 2.2.6.8 UDP-GlcNAc-2-Epimerase-Assays Zum Nachweis der UDP-GlcNAc-2-Epimerase-Aktivität werden zum einen der nichtradioaktive UDP-GlcNAc-2-Epimerase-Assay mit anschließendem colorimetrischen Nachweis des entstandenen ManNAc zum anderen der sehr viel empfindlichere radioaktive UDP-GlcNAc-2-Epimerase-Assay durchgeführt. Nichtradioaktiver UDP-GlcNAc-2-Epimerase-Assay 1: 100 μl Probe/Wasser werden mit 45 μl 200 mM Natriumphosphatpuffer, pH 7,5, 45 μl 50 mM MgCl2 und 2,5 μl 100 mM UDP-GlcNAc gemischt und in der Regel für 30 min bei 37 °C inkubiert. Die Reaktion wird durch 1-minütige Inkubation bei 100 °C gestoppt. Die denaturierten Proteine aus dem Enzymreaktionsansatz werden durch Zentrifugation bei 20000 x g für 1 min pelletiert. Das entstandene ManNAc wird mittels Morgan-Elson-Test (Reissig et [Seite 58] al., 1955), mit dem spezifisch N-Acetylhexosamine detektiert werden, nachgewiesen. Nach der Zentrifugation werden 150 μl des Überstandes mit 30 μl 0,8 M H2BO3-Puffer, pH 9,1 (mit KOH eingestellt) gemischt und für 3 min bei 100 °C inkubiert. Anschließend werden 800 μl Farbreagenz (1% (w/v) 4-Dimethylaminobenzaldehyd / 1,25% (v/v) 10 M HCl in Essigsäure) zugegeben und für 30 min bei 37 °C inkubiert. Die Extinktion wird bei 578 nm gemessen. Nichtradioaktiver UDP-GlcNAc-2-Epimerase-Assay 2: Für einige zeitabhängige Aktivitätsmessungen wird ein modifizierter UDP-GlcNAc-2-Epimerase-Assay verwendet. In diesem Epimerase-Assay 2 werden unterschiedliche Probenvolumina mit 260 μl 200 mM Tris-HCl, pH 8,1 / 65 mM MgCl2, 10 μl 100 mM UDP-GlcNAc, 40 μl frisch angesetztem 100 mM Phosphoenolpyruvat, 20 μl frisch angesetztem 15 mM NADH und 2 μl Pyruvat-Kinase/Lactat-Dehydrogenase (2 U) in einem Gesamtvolumen von 800 μl gemischt. Die Enzymreaktion wird bei 37 °C im Photometer bei 340 nm verfolgt. Als Kontrolle wird ein Ansatz ohne Enzym gemessen und die ermittelten Werte später von den Werten der Enzymreaktion abgezogen. |
Ein Verweis auf die Quelle fehlt. |
|
| [6.] Hw/Fragment 041 01 - Diskussion Bearbeitet: 31. August 2014, 12:47 Schumann Erstellt: 27. August 2014, 08:57 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 41, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 45, Zeilen: 45: 19ff; 48: 23ff |
|---|---|
| Der cytosolische Überstand wird abgenommen und für weitere Analysen verwendet. Das Pellet wird in 50 μl SDS-Probenpuffer (s. 2.2.6.3) resuspendiert.
6.2.2.4. Expression von rekombinantem Protein in S. cerevisiae Die Expressionen werden unter den ermittelten Bedingungen für eine optimale Proteinausbeute durchgeführt. Einzelne Kolonien werden in 5 ml Minimalmedium bis zur Sättigung bei 30 °C kultiviert. Mit 1 ml dieser Vorkultur werden 100 ml Minimalmedium beimpft und bis zu einer OD600 von 1 bei 30 °C kultiviert. Durch Zugabe von 2% (v/v) Galactose wird die Proteinexpression induziert. Nach 6 h werden die Hefezellen durch Zentrifugation für 5 min bei 3000 x g und 4 °C pelletiert und das Zellpellet bei -20 °C gelagert oder direkt lysiert, indem die Zellen in 1 ml Lysepuffer (10 mM Natriumphosphat, pH 7,5 / 1 mM EDTA / 1 mM DTT / 1 mM PMSF) resuspendiert werden und ein gleiches Volumen an „acid-washed 0,5 mm glass beads“ (Sigma, Deutschland) zugegeben wird. Die Zellen werden, wie unter 2.2.3.3 beschrieben, mechanisch durch Scherkräfte aufgebrochen und Zelltrümmer und unlösliche Komponenten bei 20000 x g und 4 °C für 10 min abzentrifugiert. Der cytosolische Überstand wird abgenommen und für die beschriebenen Experimente eingesetzt. 6.2.2.5. Lyse von Hefezellen Die geernteten Hefezellen von 1 ml Hefekultur werden in 100 μl Lysepuffer (10 mM Natriumphosphatpuffer, pH 7,5 / 1 mM DTT / 1 mM EDTA / 1 mM PMSF) resuspendiert und ein gleiches Volumen an „acid-washed 0,5 mm glass beads“ (Sigma, Deutschland) zugegeben. Die Zellsuspension wird für 30 sec gevortext und anschließend 30 sec auf Eis inkubiert. Dieser Vorgang wird achtmal wiederholt, damit die Zellen komplett lysiert sind. Zelltrümmer und unlösliche Komponeten werden im Anschluß bei 20000 x g und 4 °C für 30 min abzentrifugiert. Der cytosolische Überstand wird abgenommen und für weitere Analysen verwendet. Das Pellet wird in 100 μl Lysepuffer resuspendiert und den gleichen Analysen unterzogen. |
Der cytosolische Überstand wird abgenommen und für weitere Analysen verwendet. Das Pellet wird in 50 μl SDS-Probenpuffer (s. 2.2.6.3) resuspendiert.
2.2.3.4 Expression von rekombinantem Protein in Saccharomyces cerevisiae Die Expressionen werden unter den ermittelten Bedingungen für eine optimale Proteinausbeute durchgeführt. Einzelne Kolonien werden in 5 ml Minimalmedium bis zur Sättigung bei 30 °C kultiviert. Mit 1 ml dieser Vorkultur werden 100 ml Minimalmedium beimpft und bis zu einer OD600 von 1 bei 30 °C kultiviert. Durch Zugabe von 2% (v/v) Galactose wird die Proteinexpression induziert. Nach 6 h werden die Hefezellen durch Zentrifugation für 5 min bei 3000 x g und 4 °C pelletiert und das Zellpellet bei -20 °C gelagert oder direkt lysiert, indem die Zellen in 1 ml Lysepuffer (10 mM Natriumphosphat, pH 7,5 / 1 mM EDTA / 1 mM DTT / 1 mM PMSF) resuspendiert werden und ein gleiches Volumen an „acid-washed 0,5 mm glass beads“ (Sigma, Deutschland) zugegeben wird. Die Zellen werden, wie unter 2.2.3.3 beschrieben, mechanisch durch Scherkräfte aufgebrochen und Zelltrümmer und unlösliche Komponenten bei 20000 x g und 4 °C für 10 min abzentrifugiert. Der cytosolische Überstand wird abgenommen und für die beschriebenen Experimente eingesetzt. [Seite 48] 2.2.4.6 Lyse von Hefezellen Die geernteten Hefezellen von 1 ml Hefekultur werden in 100 μl Lysepuffer (10 mM Natriumphosphatpuffer, pH 7,5 / 1 mM DTT / 1 mM EDTA / 1 mM PMSF) resuspendiert und ein gleiches Volumen an „acid-washed 0,5 mm glass beads“ (Sigma, Deutschland) zugegeben. Die Zellsuspension wird für 30 sec gevortext und anschließend 30 sec auf Eis inkubiert. Dieser Vorgang wird achtmal wiederholt, damit die Zellen komplett lysiert sind. Zelltrümmer und unlösliche Komponeten werden im Anschluß bei 20000 x g und 4 °C für 30 min abzentrifugiert. Der cytosolische Überstand wird abgenommen und für weitere Analysen verwendet. Das Pellet wird in 100 μl Lysepuffer resuspendiert und den gleichen Analysen unterzogen. |
Ein Verweis auf die Quelle fehlt. Man beachte den übernommenen Verweis "s. 2.2.6.3", im ersten Satz. Dieses Unterkapitel existiert in der untersuchten Arbeit gar nicht. |
|
| [7.] Hw/Fragment 039 01 - Diskussion Bearbeitet: 31. August 2014, 12:45 Schumann Erstellt: 27. August 2014, 09:08 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 39, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 42, 44, Zeilen: 42: 9ff; 44: 1ff |
|---|---|
| [Zur Konstruktion jeder Mutante sind zwei zueinander revers] komplementäre Oligonukleotidprimer erforderlich, die die gewünschte Mutation möglichst mittig tragen. Die Primersequenzen sind unter Materialien/Primer (2.1.6) beschrieben. Als Template-DNA dient der pFASTBACHTA/2epi-Vektor, der ausgehend von den Mutationsprimern von der PfuTurbo-DNA-Polymerase repliziert wird. Die Amplifikationsreaktionen werden in einem Touch-Down-Thermocycler (Hybaid) unter folgenden Bedingungendurchgeführt [sic]:
30 sec 95 °C 16 x (30 sec 95 °C / 1 min 55 °C / 14 min 68 °C) Anschließend wird die parentale Template-DNA mit der Restriktionsendonuklease Dpn I abgebaut, die spezifisch methylierte und hemimethylierte DNA abbaut (1 h, 37 °C, 10 U). Die amplifizierte DNA mit den Mutationen wird in superkompetente Invα/F' Zellen (Invitrogen, Niederlande) transformiert. 6.2.2. Expression von rekombinantem Protein in S. cerevisiae Die Überexpression von rekombinantem Protein in der Hefe S. cerevisiae erfolgt nach den Anweisungen des Herstellers des Expressionsvektors pYES2 (Invitrogen, Niederlande). Sämtliche Expressionsexperimente werden mit dem Hefestamm INVSc1 (Invitrogen, Niederlande) durchgeführt. 6.2.2.1. Herstellung kompetenter S. cerevisiae-Hefezellen Für die Aufnahme von Fremd-DNA durch die Hefezellen werden zunächst kompetente Zellen hergestellt. Dafür werden 5 ml Vollmedium mit einer Einzelkolonie von INVSc1 angeimpft und bei 30 °C bis zur Sättigung kultiviert. 4 ml dieser Vorkultur werden in 100 ml Vollmedium übergeimpft und bis zu einer OD600 von etwa 1 angezogen. Die Zellen werden durch Zentrifugation (3000 x g, 5 min, 4 °C) sedimentiert und mit 20 ml 1 M Sorbitol / 10 mM Tricin-NaOH, pH 8,4 / 3% (v/v) Ethylenglycol gewaschen. Die sedimentierten Zellen werden in 4 ml 1 M Sorbitol / 10 mM Tricin-NaOH, pH 8,4 / 3% (v/v) Ethylenglycol resuspendiert und mit 100 μl zerschallter Heringssperma-DNA (10 mg/ml, zerschallt auf eine mittlere Fragmentgröße von 7 kb) sowie mit 100 μl einer sterilfiltrierten 1 M Histamin-Lösung versetzt. Aliquots von 200 μl werden bei -70 °C für mindestens 2 h eingefroren. |
Zur Konstruktion jeder Mutante sind zwei zueinander revers komplementäre Oligonukleotidprimer erforderlich, die die gewünschte Mutation möglichst mittig tragen. Die Primersequenzen sind unter Materialien/Primer (2.1.6) beschrieben. Als Template-DNA dient der pFASTBACHTA/2epi-Vektor, der ausgehend von den Mutationsprimern von der PfuTurbo-DNA-Polymerase repliziert wird. Die Amplifikationsreaktionen werden in einem Touch-Down-Thermocycler (Hybaid) unter folgenden Bedingungen durchgeführt:
30 sec 95 °C 16 x (30 sec 95 °C / 1 min 55 °C / 14 min 68 °C) Anschließend wird die parentale Template-DNA mit der Restriktionsendonuklease Dpn I abgebaut, die spezifisch methylierte und hemimethylierte DNA abbaut (1 h, 37 °C, 10 U). Die amplifizierte DNA mit den Mutationen wird in superkompetente InvαF' Zellen (Invitrogen, Niederlande) transformiert. [Seite 44] 2.2.3 Expression von rekombinantem Protein in Saccharomyces cerevisiae Die Überexpression von rekombinantem Protein in der Hefe S. cerevisiae erfolgt nach den Anweisungen des Herstellers des Expressionsvektors pYES2 (Invitrogen, Niederlande). Sämtliche Expressionsexperimente werden mit dem Hefestamm INVSc1 (Invitrogen, Niederlande) durchgeführt. 2.2.3.1 Herstellung kompetenter Saccharomyces cerevisiae-Hefezellen Für die Aufnahme von Fremd-DNA durch die Hefezellen werden zunächst kompetente Zellen hergestellt, d.h. Zellen, die fähig sind, freie, zirkuläre DNA aufzunehmen. Dafür werden 5 ml Vollmedium mit einer Einzelkolonie von INVSc1 angeimpft und bei 30 °C bis zur Sättigung kultiviert. 4 ml dieser Vorkultur werden in 100 ml Vollmedium übergeimpft und bis zu einer OD600 von etwa 1 angezogen. Die Zellen werden durch Zentrifugation (3000 x g, 5 min, 4 °C) sedimentiert und mit 20 ml 1 M Sorbitol / 10 mM Tricine-NaOH, pH 8,4 / 3% (v/v) Ethylenglycol gewaschen. Die sedimentierten Zellen werden in 4 ml 1 M Sorbitol / 10 mM Tricine-NaOH, pH 8,4 / 3% (v/v) Ethylenglycol resuspendiert und mit 100 μl zerschallter Heringssperma-DNA (10 mg/ml, zerschallt auf eine mittlere Fragmentgröße von 7 kb) sowie mit 100 μl einer sterilfiltrierten 1 M Histamin-Lösung versetzt. Aliquots von 200 μl werden bei -70 °C für mindestens 2 h eingefroren. |
Ein Verweis auf die Quelle fehlt. |
|
| [8.] Hw/Fragment 038 01 - Diskussion Bearbeitet: 31. August 2014, 12:44 Schumann Erstellt: 27. August 2014, 09:14 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 39, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 41, 42, Zeilen: 41: 4ff; 42: 1ff |
|---|---|
| [Bei der Sequenzierung mit Fluoreszenz-markierten Sequenzierprimern werden 4 pmol Sequenzierprimer mit 1,2 μg DNA auf vier Nukleotidmixe („Thermo SequenaseTM Primer Cycle Sequencing Kit“; Amersham, Deutschland) verteilt und die] Ansätze mit einem Gesamtvolumen von 6 μl einem Cycle-Sequencing mit 25 Zyklen (20 sec 95°C / 20 sec 60 °C / 10 sec 72°C) unterzogen. Anschließend wird 1 μl des zuvor mit 4 μl Gelladepuffer gestoppten Reaktionsmixes auf ein 6%iges Polyacrylamid-Gel aufgetragen und in einerautomatischen [sic] Sequenziereinrichtung (LI-COR 4200; MWG-Biotech, Deutschland) analysiert.
6.2.1.12. Polymerase-Ketten-Reaktion (PCR) Mit Hilfe der Polymerase-Ketten-Reaktion (PCR) kann eine ausgewählte DNA-Sequenz in vitro exponentiell amplifiziert werden, z.B. um die Anwesenheit eines bestimmten DNA-Fragments nachzuweisen. Zur Synthese werden eine DNA-Sequenz, die als Template bezeichnet wird, und zwei Oligonukleotidprimer benötigt, die die zu amplifizierende Region flankieren. Die PCR wird mit dem „AmpliTaq DNA Polymerase with GeneAmp Kit“ (Perkin Elmer, USA) nach Anweisungen des Herstellers durchgeführt. Ein Standardansatz für eine PCR sieht dabei wie folgt aus: Template 10-100 ng Primer je 20 pmol dNTPs je 2 mM MgCl2 1,5 mM Puffer 10 x PCR-Buffer II Taq-Polymerase 0,5 μl/50 μl Ansatz Die Reaktion wird in einem Touch-Down-Thermocycler (Hybaid) mit folgendem Programm durchgeführt: 3 min 95 °C 30 x (30 sec 95 °C / 30 sec 55 °C / 2 min 72 °C) 7 min 72 °C Ein Aliquot von 5 μl wird zur Analyse auf ein Agarosegel aufgetragen. 6.2.1.13. Herstellung von Punktmutanten Zur Generierung von Mutanten wird der „QuickChangeTM Site-Directed-Mutagenesis Kit“ von Stratagene (USA) eingesetzt. |
Bei der Sequenzierung mit Fluoreszenz-markierten Sequenzierprimern werden 4 pmol Sequenzierprimer mit 1,2 μg DNA auf vier Nukleotidmixe („Thermo SequenaseTM Primer Cycle Sequencing Kit“; Amersham, Deutschland) verteilt und die Ansätze mit einem Gesamtvolumen von 6 μl einem Cycle-Sequencing mit 25 Zyklen (20sec 95°C / 20 sec 60 °C / 10sec 72°C) unterzogen. Anschließend wird 1 μl des zuvor mit 4 μl Gelladepuffer gestoppten Reaktionsmixes auf ein 6%iges Polyacrylamid-Gel aufgetragen und in einer automatischen Sequenziereinrichtung (LI-COR 4200; MWG-Biotech, Deutschland) analysiert.
[...] 2.2.1.13 Polymerase-Ketten-Reaktion (PCR) Mit Hilfe der Polymerase-Ketten-Reaktion (PCR) kann eine ausgewählte DNA-Sequenz in vitro exponentiell amplifiziert werden, z.B. um die Anwesenheit eines bestimmten DNA-Fragments nachzuweisen. Zur Synthese werden eine DNA-Sequenz, die als Template bezeichnet wird, und zwei Oligonukleotidprimer benötigt, die die zu amplifizierende Region flankieren. Die PCR wird mit dem „AmpliTaq®DNA Polymerase with GeneAmp® Kit“ (Perkin Elmer, USA) nach Anweisungen des Herstellers durchgeführt. Ein Standardansatz für eine PCR sieht dabei wie folgt aus:
[Seite 42] Die Reaktion wird in einem Touch-Down-Thermocycler (Hybaid) mit folgendem Programm durchgeführt:
Ein Aliquot von 5 μl wird zur Analyse auf ein Agarosegel aufgetragen. 2.2.1.14 Herstellung von Punktmutanten Zur Generierung von Mutanten wird der „QuickChangeTM Site-Directed-Mutagenesis Kit“ von Stratagene (USA) eingesetzt. |
Ein Verweis auf die Quelle fehlt. |
|
| [9.] Hw/Fragment 037 01 - Diskussion Bearbeitet: 31. August 2014, 12:43 Schumann Erstellt: 27. August 2014, 12:14 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 37, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 40, 41, Zeilen: 40: 14ff; 41: 1ff |
|---|---|
| 6.2.1.9. Herstellung kompetenter E. coli-Zellen
Kompetente Zellen sind Zellen, die fähig sind, freie, zirkuläre DNA aufzunehmen. Durch Behandlung mit CaCl2 wird die Zellmembran für DNA-Moleküle durchlässig (Dagert und Ehrlich, 1979). Zur Herstellung von kompetenten Zellen werden 2 ml LBMedium mit einer Einzelkolonie von Inv/F' angeimpft und bei 37 °C bis zur Sättigung kultiviert. 1 ml dieser Vorkultur wird in 100 ml LB überimpft und bis zu einer OD600 von 0,2-0,3 angezogen. Die Zellen werden durch Zentrifugation (3000 x g, 5 min, 4 °C) sedimentiert und in 20 ml kaltem 100 mM CaCl2 resuspendiert. Nach 30-minütiger Inkubation auf Eis wird 5 min bei 3000 x g und 4 °C zentrifugiert, das Zellpellet in 1 ml kaltem 100 mM CaCl2 resuspendiert und nochmals für mindestens 1 h auf Eis inkubiert. Die Zellen werden direkt zur Transformation eingesetzt oder nach Zugabe von 20% (v/v) Glycerin bei –80 °C gelagert. 6.2.1.10. Transformation von Plasmid-DNA in Escherichia coli Als Transformation wird die Aufnahme von Fremd-DNA und somit die genetische Veränderung von Bakterien bezeichnet. Dabei wird zu 100 μl kompetenten Zellen der Ligationsansatz oder etwa 100 ng Plasmid-DNA pipettiert und vorsichtig gemischt. Die Proben werden anschließend 30 min auf Eis inkubiert. Durch die nachfolgende 45-sec- Inkubation bei 42 °C im Wasserbad wird die Aufnahme der DNA durch die Bakterienzellen erleichtert. Die Zellen werden anschließend nochmals 2 min auf Eis inkubiert und danach in 250 μl LB-Medium oder alternativ SOC-Medium für 1 h schüttelnd bei 37 °C inkubiert, damit sich die Bakterienzellen regenerieren können. Aliquots von 50 bis 200 μl werden auf vorgewärmten Agarplatten mit Selektivmedium ausplattiert und über Nacht bei 37 °C inkubiert. 6.2.1.11. Sequenzanalyse Zur Kontrolle von Klonierungsschritten und zur Sequenzanalyse generell werden Sequenzierungen durchgeführt. Alle Sequenzierungen werden nach der Kettenabbruchmethode (Sanger et al., 1992) durchgeführt. Bei der Sequenzierung mit Fluoreszenz-markierten Sequenzierprimern werden 4 pmol Sequenzierprimer mit 1,2 μg DNA auf vier Nukleotidmixe („Thermo SequenaseTM Primer Cycle Sequencing Kit“; Amersham, Deutschland) verteilt und die [Ansätze mit einem Gesamtvolumen von 6 μl einem Cycle-Sequencing mit 25 Zyklen (20 sec 95°C / 20 sec 60 °C / 10 sec 72°C) unterzogen.] |
2.2.1.10 Herstellung kompetenter Escherichia coli-Zellen
Kompetente Zellen sind Zellen, die fähig sind, freie, zirkuläre DNA aufzunehmen. Durch Behandlung mit CaCl2 wird die Zellmembran für DNA-Moleküle durchlässig (Dagert und Ehrlich, 1979). Zur Herstellung von kompetenten Zellen werden 2 ml LBMedium mit einer Einzelkolonie von InvαF' oder BL21 StarTM(DE3)pLysS angeimpft und bei 37 °C bis zur Sättigung kultiviert. 1 ml dieser Vorkultur wird in 100 ml LB überimpft und bis zu einer OD600 von 0,2-0,3 angezogen. Die Zellen werden durch Zentrifugation (3000 x g, 5 min, 4 °C) sedimentiert und in 20 ml kaltem 100 mM CaCl2 resuspendiert. Nach 30-minütiger Inkubation auf Eis wird 5 min bei 3000 x g und 4 °C zentrifugiert, das Zellpellet in 1 ml kaltem 100 mM CaCl2 resuspendiert und nochmals für mindestens 1 h auf Eis inkubiert. Die Zellen werden direkt zur Transformation eingesetzt oder nach Zugabe von 20% (v/v) Glycerin bei –80 °C gelagert. 2.2.1.11 Transformation von Plasmid-DNA in Escherichia coli Als Transformation wird die Aufnahme von Fremd-DNA und somit die genetische Veränderung von Bakterien bezeichnet. Dabei wird zu 100 μl kompetenten Zellen der Ligationsansatz oder etwa 100 ng Plasmid-DNA pipettiert und vorsichtig gemischt. Die Proben werden anschließend 30 min auf Eis inkubiert. Durch die nachfolgende 45-sec- Inkubation bei 42 °C im Wasserbad wird die Aufnahme der DNA durch die Bakterienzellen erleichtert. Die Zellen werden anschließend nochmals 2 min auf Eis inkubiert und danach in 250 μl LB-Medium oder alternativ SOC-Medium für 1 h schüttelnd bei 37 °C inkubiert, damit sich die Bakterienzellen regenerieren können. Aliquots von 50 bis 200 μl werden auf vorgewärmten Agarplatten mit Selektivmedium ausplattiert und über Nacht bei 37 °C inkubiert. [Seite 41] 2.2.1.12 Sequenzierungen Zur Kontrolle von Klonierungsschritten und zur Sequenzanalyse generell werden Sequenzierungen durchgeführt. Alle Sequenzierungen werden nach der Kettenabbruchmethode (Sanger et al., 1992) durchgeführt. Bei der Sequenzierung mit Fluoreszenz-markierten Sequenzierprimern werden 4 pmol Sequenzierprimer mit 1,2 μg DNA auf vier Nukleotidmixe („Thermo SequenaseTM Primer Cycle Sequencing Kit“; Amersham, Deutschland) verteilt und die Ansätze mit einem Gesamtvolumen von 6 μl einem Cycle-Sequencing mit 25 Zyklen (20sec 95°C / 20 sec 60 °C / 10sec 72°C) unterzogen. |
Ein Verweis auf die Quelle fehlt. |
|
| [10.] Hw/Fragment 036 01 - Diskussion Bearbeitet: 31. August 2014, 12:41 Schumann Erstellt: 27. August 2014, 12:17 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 36, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 39, 40, Zeilen: 39: 17ff - 40: 1ff |
|---|---|
| [Gleichzeitig führen die im Puffer enthaltenen Salze] zu einer leichten Modifikation der Nucleinsäure-Struktur, so daß anschließend eine optimale Bindung an die Silicagel-Membran der Säulen möglich ist. Die DNA-Bindung an das Säulenmaterial ermöglicht die Reinigung von unerwünschten Primern und Verunreinigungen, wie Salzen, Enzymen, nicht inkorporierte Nucleotiden, Agarose, Ethidiumbromid, Ölen oder Detergenzien durch den ethanolhaltigen PE-Puffer. Die Elution erfolgt anschließend mit 30 μl Wasser. Die Isolierung von DNA-Fragmenten aus Agarosegelen wird entsprechend den Anweisungen des Herstellers durchgeführt.
6.2.1.7. Ligation von DNA-Fragmenten Die Verknüpfung von DNA-Fragmenten mit überhängenden oder glatten Enden mittels einer Ligase wird als Ligation bezeichnet. Bei der Standardligation wird durch Bildung von Phosphodiesterbindungen zwischen benachbarten 3'-Hydroxyl- und 5'-Phosphatgruppen linearisierter Vektor mit isolierten DNA-Fragmenten verknüpft, die durch Restriktionsspaltung und anschließende Gelelektrophorese mit Gelelution isoliert worden sind. Für die Ligation werden Vektor und Insert im Verhältnis von mindestens 1:3 eingesetzt. Der Ansatz wird mit 1 μl T4-DNA Ligase (20 U; GibcoBRL, USA) pro 10 μl Ansatz bei RT für 5-15 min inkubiert und anschließend vollständig zur Transformation von Bakterien eingesetzt. 6.2.1.8. Dephosphorylierung von DNA Linearisierte Vektoren, deren Enden symmetrisch sind, zeigen eine hohe Religationsrate. Um bei Religationen mit DNA-Fragmenten die Selbstligationsrate des Vektors zu verringern, wird vorher das linearisierte Plasmid mit bakterieller alkalischer Phosphatase aus E. coli behandelt. Diese alkalische Phosphatase katalysiert die Abspaltung der 5'-terminalen Phosphatgruppe von DNA, die bei Ligationen als Energielieferant benötigt wird, wodurch die Religationsrate des Vektors stark verringert wird. Die Dephosphorylierung der DNA wird entsprechend den Anweisungen des Herstellers (GibcoBRL, USA) durchgeführt. Die in 100 μl 10 mM Tris-HCl, pH 8,0, gelöste DNA (1 pmol) wird nach der Zugabe von 1 μl bakterieller alkalischer Phosphatase 1 h bei 65 °C inkubiert. Anschließend wird die DNA mit dem „QIAquick PCR Purification Kit“ (Qiagen, Deutschland) gereinigt und in 20 μl Wasser aufgenommen. |
Gleichzeitig führen die im Puffer enthaltenen Salze zu einer leichten Modifikation der Nukleinsäure- Struktur, so daß anschließend eine optimale Bindung an die Silicagel-Membran der Säulen möglich ist. Die DNA-Bindung an das Säulenmaterial ermöglicht die Reinigung von unerwünschten Primern und Verunreinigungen, wie Salzen, Enzymen, nicht inkorporierte Nucleotiden, Agarose, Ethidiumbromid, Ölen oder Detergenzien durch den ethanolhaltigen PE-Puffer. Die Elution erfolgt anschließend mit 30 μl Wasser. Die Isolierung von DNA-Fragmenten aus Agarosegelen wird entsprechend den Anweisungen des Herstellers durchgeführt.
2.2.1.8 Ligation von DNA-Fragmenten Die Verknüpfung von DNA-Fragmenten mit überhängenden oder glatten Enden mittels einer Ligase wird als Ligation bezeichnet. Bei der Standardligation wird durch Bildung von Phosphodiesterbindungen zwischen benachbarten 3'-Hydroxyl- und 5'-Phosphatgruppen linearisierter Vektor mit isolierten DNA-Fragmenten verknüpft, die durch Restriktionsspaltung und anschließende Gelelektrophorese mit Gelelution isoliert worden sind. Für die Ligation werden Vektor und Insert im Verhältnis von mindestens 1:3 eingesetzt. Der Ansatz wird mit 1 μl T4-DNA Ligase (20 U; GibcoBRL, USA) pro 10 μl Ansatz bei RT für 5-15 min inkubiert und anschließend vollständig zur Transformation von Bakterien eingesetzt. [Seite 40] 2.2.1.9 Dephosphorylierung von DNA Linearisierte Vektoren, deren Enden symmetrisch sind, zeigen eine hohe Religationsrate. Um bei Religationen mit DNA-Fragmenten die Selbstligationsrate des Vektors zu verringern, wird vorher eine Behandlung des linearisierten Plasmids mit Bakterieller-Alkalischer-Phosphatase aus E. coli durchgeführt. Diese Alkalische Phosphatase katalysiert die Abspaltung der 5'-terminalen Phosphatgruppe von DNA, die bei Ligationen als Energielieferant benötigt wird, wodurch die Religationsrate des Vektors stark verringert wird. Die Dephosphorylierung der DNA wird entsprechend den Anweisungen des Herstellers (GibcoBRL, USA) durchgeführt. Die in 100 μl 10 mM Tris-HCl, pH 8,0 gelöste DNA (1 pmol) wird nach der Zugabe von 1 μl Bakterieller- Alkalischer-Phosphatase 1 h bei 65 °C inkubiert. Anschließend wird die DNA mit dem „QIAquick PCR Purification Kit“ (Qiagen, Deutschland) gereinigt und in 20 μl Wasser aufgenommen. |
Ein Verweis auf die Quelle fehlt. |
|
| [11.] Hw/Fragment 035 01 - Diskussion Bearbeitet: 31. August 2014, 12:40 Schumann Erstellt: 27. August 2014, 12:20 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 35, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 38, 39, Zeilen: 38: 18f - 39:1ff |
|---|---|
| [Ein Reaktionsansatz enthält zwischen 1 und 40 μg DNA. Pro Restriktionsansatz] werden etwa eine Einheit (U) Enzym pro μg DNA eingesetzt. Anschließend werden die erhaltenen Fragmente mit Hilfe der Agarose-Gelelektrophorese analysiert.
6.2.1.5. Agarose-Gelelektrophorese Mit der Agarose-Gelelektrophorese können DNA-Moleküle nach ihrer Größe aufgetrennt werden. Die Wanderungsgeschwindigkeit linearer DNA-Moleküle ist dabei umgekehrt propotional zum Logarithmus ihres Molekulargewichtes. Bei ringförmiger DNA können auch coiled- und supercoiled-Strukturen auftreten, welche aufgrund ihrer höheren Kompaktheit schneller wandern und in den Gelen deshalb bei scheinbar kleineren Molekulargewichten detektiert werden. Mit Agarosegelen können DNA-Moleküle im Bereich von 500-8000 bp identifiziert werden. Agarose-Gelelektrophoresen werden zur Größenbestimmung von DNA-Fragmenten nach Restriktionsspaltung, zur Isolierung von DNA-Fragmenten und zur Qualitätskontrolle von DNA verwendet. Die elektrophoretische Auftrennung erfolgt in horizontalen 0,8 bis 1,5%igen (w/v) Agarose-Gelen. Die Agarose wird durch Kochen in TAE-Puffer (40 mM Tris-HCl / 5 mM Natriumacetat / 1 mM EDTA, pH 7,9) gelöst und in die entsprechenden Gelschlitten gegossen. Nach dem Abkühlen und Erstarren der Agarose wird das Gel in die mit TAE-Puffer gefüllte Elektrophoresekammer gelegt. Die Proben werden vor dem Auftragen mit 1/5 Volumen Bromphenolblau-Probenpuffer (20% (w/v) Ficoll 400 / 1% (w/v) SDS / 0,02% (w/v) Bromphenolblau in TAE-Puffer) versetzt. Das Auftragsvolumen beträgt etwa 5-20 μl pro Probentasche. Als Größenstandard werden 10 μl des 1 kb-DNA-Markers (Gibco- BRL, USA) aufgetragen. Die Elektrophorese wird anschließend bei 0,5 V / cm2 durchgeführt, bis der Farbmarker die gewünschte Laufstrecke zurückgelegt hat. Das Gel wird dann für 5-10 min in einem Ethidiumbromidbad (0,5 μg/ml Ethidiumbromid in TAE-Puffer) inkubiert und die DNA unter UV-Licht (254 nm) sichtbar gemacht. 6.2.1.6. Isolierung von DNA-Fragmenten mittels Gelelution DNA kann aus Agarose-Gelen durch Verflüssigung der Agarose in Spezialpuffer herausgelöst und anschließend über QIAgen-Säulen des „QlAquick Gel Extraction Kits“ (Qiagen, Deutschland) gereinigt werden. Der im Kit enthaltene Puffer QG ermöglicht das Schmelzen der Agarose bei relativ niedrigen Temperaturen. |
Ein Reaktionsansatz enthält zwischen 1 und 40 μg DNA. Pro Restriktionsansatz werden etwa eine Einheit (U) Enzym pro μg DNA eingesetzt. Anschließend werden die erhaltenen Fragmente mit Hilfe der Agarose-Gelelektrophorese analysiert.
2.2.1.6 Agarose-Gelelektrophorese Mit der Agarose-Gelelektrophorese können DNA-Moleküle nach ihrer Größe aufgetrennt werden. Die Wanderungsgeschwindigkeit linearer DNA-Moleküle ist dabei umgekehrt propotional zum Logarithmus ihres Molekulargewichtes. Bei ringförmiger DNA können auch coiled und supercoiled-Strukturen auftreten, welche aufgrund ihrer höheren Kompaktheit schneller wandern und in den Gelen deshalb bei scheinbar kleineren Molekulargewichten detektiert werden. Mit Agarosegelen können DNA-Moleküle im Bereich von 500-8000 bp identifiziert werden. Agarose-Gelelektrophoresen werden zur Größenbestimmung von DNA-Fragmenten nach Restriktionsspaltung, zur Isolierung von DNA-Fragmenten und zur Qualitätskontrolle von DNA verwendet. Die elektrophoretische Auftrennung erfolgt in [Seite 39] horizontalen 0,8 bis 1,5%igen (w/v) Agarose-Gelen. Die Agarose wird durch Kochen in TAE-Puffer (40 mM Tris / 5 mM Natriumacetat / 1 mM EDTA, pH 7,9) gelöst und in die entsprechenden Gelschlitten gegossen. Nach dem Abkühlen und Erstarren der Agarose wird das Gel in die mit TAE-Puffer gefüllte Elektrophoresekammer gelegt. Die Proben werden vor dem Auftragen mit 1/5 Volumen Bromphenolblau-Probenpuffer (20% (w/v) Ficoll 400 / 1% (w/v) SDS / 0,02% (w/v) Bromphenolblau in TAE-Puffer) versetzt. Das Auftragsvolumen beträgt etwa 5-20 μl pro Probentasche. Als Größenstandard werden 10 μl des 1 kb-DNA-Markers (Gibco-BRL, USA) aufgetragen. Die Elektrophorese wird anschließend bei 0,5 V / cm2 durchgeführt, bis der Farbmarker die gewünschte Laufstrecke zurückgelegt hat. Das Gel wird dann für 5-10 min in einem Ethidiumbromidbad (0,5 μg/ml Ethidiumbromid in TAE-Puffer) inkubiert und die DNA unter UV-Licht (254 nm) sichtbar gemacht. 2.2.1.7 Isolierung von DNA-Fragmenten mittels Gelelution DNA kann aus Agarose-Gelen durch Verflüssigung der Agarose in Spezialpuffer herausgelöst und anschließend über QIAgen-Säulen des „QlAquick Gel Extraction Kits“ (Qiagen, Deutschland) gereinigt werden. Der im Kit enthaltene Puffer QG ermöglicht das Schmelzen der Agarose bei relativ niedrigen Temperaturen. |
Ein Verweis auf die Quelle fehlt. |
|
| [12.] Hw/Fragment 034 01 - Diskussion Bearbeitet: 31. August 2014, 12:38 Schumann Erstellt: 27. August 2014, 12:24 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 34, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 36, 37, Zeilen: 36: letzte Zeile - 37: 1-2, 11ff |
|---|---|
| Das Präzipitat wird anschließend getrocknet, in 60 μl bzw. 150 μl Wasser oder TE-Puffer (10 mM Tris-HCl, pH 7,5 / 1 mM EDTA) aufgenommen und quantifiziert bzw. analysiert.
6.2.1.2. Fällung von DNA DNA läßt sich durch Ethanol oder 2-Propanol fällen. Dafür werden 2 Volumina 2-Propanol bzw. 1/10 Volumen 3 M Natriumacetat-Puffer, pH 4,8, und 2,5 Volumina Ethanol zugegeben, der Ansatz durch vortexen gut gemischt und 30 min bei –20 °C inkubiert. Die gefällte DNA wird anschließend 10 min bei 20000 x g pelletiert und das Pellet mit kaltem 70% (v/v) Ethanol gewaschen. Das trockene Pellet wird in 5-20 μl Wasser oder TE-Puffer (10 mM Tris-HCl, pH 7,5 / 1 mM EDTA) aufgenommen. 6.2.1.3. Bestimmung von DNA-Konzentrationen DNA-Konzentrationen werden spektralphotometrisch durch Absorptionsmessungen bei 260 nm ermittelt. Ein Extinktionswert von 1,0 entspricht dabei 50 μg Doppelstrang-DNA/ml Lösung. Wird gleichzeitig die Absorption bei 280 nm gemessen, dem Absorptionsmaximum von Proteinen, so kann aus dem Verhältnis 260 nm zu 280 nm die Reinheit der DNA-Probe bestimmt werden. Saubere DNA ist frei von Proteinen und RNA und hat ein Verhältnis der Wellenlängen 260 nm / 280 nm von ca. 1,8. Eine weitere Möglichkeit DNA-Mengen zu quantifizieren, besteht in der Abschätzung der Bandenintensität auf Agarosegelen, im Vergleich zu bekannten DNA-Mengen. 6.2.1.4. DNA-Spaltungen mit Restriktionsendonukleasen Restriktionsendonukleasen erkennen spezifische Sequenzen von 4-8 bp und führen in doppelsträngige DNA Strangbrüche ein, wobei überhängende 3' bzw. 5' Enden (sticky ends) oder glatte Enden (blunt end) entstehen. Die Spaltung von DNA-Doppelsträngen mit Restriktionsendonukleasen wird für die Analyse, Klonierung und Fragmentisolierung von DNA eingesetzt. Restriktionsspaltungen werden unter den vom Hersteller empfohlenen Bedingungen durchgeführt. Ein Reaktionsansatz enthält zwischen 1 und 40 μg DNA. |
[Seite 36]
Das [Seite 37] Präzipitat wird anschließend getrocknet, in 60 μl bzw. 150 μl Wasser oder TE-Puffer (10 mM Tris-HCl, pH 7,5 / 1 mM EDTA) aufgenommen und quantifiziert bzw. analysiert. [...] 2.2.1.3 Fällung von DNA DNA läßt sich durch Ethanol oder 2-Propanol fällen. Dafür werden 2 Volumina 2- Propanol bzw. 1/10 Volumen 3 M Natriumacetat-Puffer, pH 4,8 und 2,5 Volumina Ethanol zugegeben, der Ansatz durch vortexen gut gemischt und 30 min bei –20 °C inkubiert. Die gefällte DNA wird anschließend 10 min bei 20000 x g pelletiert und das [Seite 38] Pellet mit kaltem 70% (v/v) Ethanol gewaschen. Das trockene Pellet wird in 5-20 μl Wasser oder TE-Puffer (10 mM Tris-HCl, pH 7,5 / 1 mM EDTA) aufgenommen. 2.2.1.4 Bestimmung von DNA-Konzentrationen DNA-Konzentrationen werden spektralphotometrisch durch Absorptionsmessungen bei 260 nm ermittelt. Ein Extinktionswert von 1,0 entspricht dabei 50 μg Doppelstrang- DNA/ml Lösung. Wird gleichzeitig die Absorption bei 280 nm gemessen, dem Absorptionsmaximum von Proteinen, so kann aus dem Verhältnis 260 nm zu 280 nm die Reinheit der DNA-Probe bestimmt werden. Saubere DNA ist frei von Proteinen und RNA und hat ein Verhältnis der Wellenlängen 260 nm / 280 nm von ca. 1,8. Eine weitere Möglichkeit DNA-Mengen zu quantifizieren, besteht in der Abschätzung der Bandenintensität auf Agarosegelen, im Vergleich zu bekannten DNA-Mengen. 2.2.1.5 DNA-Spaltungen mit Restriktionsendonukleasen Restriktionsendonukleasen erkennen spezifische Sequenzen von 4-8 bp und führen in doppelsträngige DNA Strangbrüche ein, wobei überhängende 3' bzw. 5' Enden (sticky ends) oder glatte Enden (blunt end) entstehen. Die Spaltung von DNA-Doppelsträngen mit Restriktionsendonukleasen wird für die Analyse, Klonierung und Fragmentisolierung von DNA eingesetzt. Restriktionsspaltungen werden unter den vom Hersteller empfohlenen Bedingungen durchgeführt. Ein Reaktionsansatz enthält zwischen 1 und 40 μg DNA. |
Ein Verweis auf die Quelle fehlt. |
|
| [13.] Hw/Fragment 033 01 - Diskussion Bearbeitet: 31. August 2014, 12:37 Schumann Erstellt: 27. August 2014, 12:28 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 33, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 36, 37, Zeilen: 36: 17ff - 37: 1ff |
|---|---|
| Zentrifuge Megafuge 1.0 Heraeus
6.2. Methoden 6.2.1. Allgemeine molekularbiologische Methoden 6.2.1.1. Präparation von Plasmid-DNA Alle Plasmidpräparationen werden mit dem „Plasmid Purification Kit“ der Firma Qiagen (Deutschland) durchgeführt. Die Methode geht auf die alkalische Lyse nach Birnboim und Doly (1979) zurück. Für eine Mini-Plasmidpräparation (30-50 μg DNA) werden von einer E. coliÜbernachtkultur in selektivem LB-Medium 2 ml in ein Eppendorf-Reaktionsgefäß überführt und 10 min bei 20000 x g zentrifugiert. Das erhaltene Bakterienpellet wird in 200 μl Puffer I, versetzt mit 100 mg/l RNase A, unter leichtem Vortexen gut resuspendiert. Anschließend werden 200 μl Puffer II zugegeben, durch vorsichtiges Schwenken wird der Ansatz gut gemischt und 5 min bei RT inkubiert. Die stark alkalische Pufferlösung II lysiert die Zellen und setzt dadurch die Plasmid-DNA frei. Nach Zugabe von 200 μl des Neutralisations-Puffers III wird der Ansatz mindestens 15 min auf Eis inkubiert. Durch die hohe Salzkonzentration dieser Lösung werden die Proteine und die chromosomale DNA gefällt, während die Plasmid-DNA gelöst bleibt. Durch eine 15-minütige Zentrifugation bei 20000 x g werden die zellulären Proteine und die chromosomale DNA pelletiert. Der Überstand mit der Plasmid-DNA wird nochmals für 15 min zentrifugiert. Anschließend wird die Plasmid-DNA im Überstand mit 450 μl (0,7 Volumen) 2- Propanol gefällt und nach 30-minütiger Inkubation bei –20 °C durch Zentrifugation für 15 min bei 20000 x g pelletiert. Um restliche Salze zu entfernen, wird das DNA-Pellet mit 500 μl kaltem 70% (v/v) Ethanol gewaschen. Das Präzipitat wird anschließend getrocknet, in 30 μl Wasser oder TE-Puffer (10 mM Tris- HCl, pH 7,5 / 1 mM EDTA) aufgenommen und quantifiziert bzw. analysiert. Um größere Mengen (100 bzw. 500 μg) High-Copy-Plasmid-DNA zu isolieren, werden 25 ml bzw. 100 ml E. coli-Übernachtkulturen angesetzt und das Plasmid wie oben beschrieben isoliert. Bevor die Plasmid-DNA mit 2-Propanol gefällt wird, erfolgt bei diesen Midi- bzw. Maxi-Plasmidpräparationen eine weitere Reinigung der DNA über ein Anionen- Austauscher-Harz, an das Plasmid-DNA bei geringen Salzkonzentrationen und niedrigem pH-Wert bindet. Verunreinigungen, wie z.B. Proteine, werden bei mittleren Salzkonzentrationen entfernt. Mit Hochsalzpuffer wird die Plasmid-DNA eluiert, durch 2-Propanol gefällt und mit [kaltem 70% (v/v) Ethanol gewaschen, um restliche Salze zu entfernen.] |
Zentrifuge Megafuge 1.0 Heraeus
2.2. Methoden 2.2.1 Allgemeine molekularbiologische Methoden 2.2.1.1 Präparation von Plasmid-DNA Alle Plasmidpräparationen werden mit dem „Plasmid Purification Kit“ der Firma Qiagen (Deutschland) durchgeführt. Die Methode geht auf die alkalische Lyse nach Birnboim und Doly (1979) zurück. Für eine Mini-Plasmidpräparation (30-50 μg DNA) werden von einer E. coliÜbernachtkultur in selektivem LB-Medium 2 ml in ein Eppendorf-Reaktionsgefäß überführt und 10 min bei 20000 x g zentrifugiert. Das erhaltene Bakterienpellet wird in 200 μl Puffer I, versetzt mit 100 mg/l RNase A, unter leichtem Vortexen gut resuspendiert. Anschließend werden 200 μl Puffer II zugegeben, durch vorsichtiges Schwenken wird der Ansatz gut gemischt und 5 min bei RT inkubiert. Die stark alkalische Pufferlösung II lysiert die Zellen und setzt dadurch die Plasmid-DNA frei. Nach Zugabe von 200 μl des Neutralisations-Puffers III wird der Ansatz mindestens 15 min auf Eis inkubiert. Durch die hohe Salzkonzentration dieser Lösung werden die Proteine und die chromosomale DNA gefällt, während die Plasmid-DNA gelöst bleibt. Durch eine 15-minütige Zentrifugation bei 20000 x g werden die zellulären Proteine und die chromosomale DNA pelletiert. Der Überstand mit der Plasmid-DNA wird nochmals für 15 min zentrifugiert. Anschließend wird die Plasmid-DNA im Überstand mit 450 μl (0,7 Volumen) 2-Propanol gefällt und nach 30-minütiger Inkubation bei –20 °C durch Zentrifugation für 15 min bei 20000 x g pelletiert. Um restliche Salze zu entfernen, wird das DNA-Pellet mit 500 μl kaltem 70% (v/v) Ethanol gewaschen. Das [Seite 38] Präzipitat wird anschließend getrocknet, in 30 μl Wasser oder TE-Puffer (10 mM Tris- HCl, pH 7,5 / 1 mM EDTA) aufgenommen und quantifiziert bzw. analysiert. Um größere Mengen (100 bzw. 500 μg) High-Copy-Plasmid-DNA zu isolieren, werden 25 ml bzw. 100 ml E. coli-Übernachtkulturen angesetzt und das Plasmid wie oben beschrieben isoliert. Bevor die Plasmid-DNA mit 2-Propanol gefällt wird, erfolgt bei diesen Midi- bzw. Maxi-Plasmidpräparationen eine weitere Reinigung der DNA über ein Anionen-Austauscher-Harz, an das Plasmid-DNA bei geringen Salzkonzentrationen und niedrigem pH-Wert bindet. Verunreinigungen, wie z.B. Proteine, werden bei mittleren Salzkonzentrationen entfernt. Mit Hochsalzpuffer wird die Plasmid-DNA eluiert, durch 2-Propanol gefällt und mit kaltem 70% (v/v) Ethanol gewaschen, um restliche Salze zu entfernen. |
Ein Verweis auf die Quelle fehlt. |
|
| [14.] Hw/Fragment 030 01 - Diskussion Bearbeitet: 31. August 2014, 12:35 Schumann Erstellt: 27. August 2014, 12:37 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 30, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 32, Zeilen: 32: 1ff; 33: 1-2 |
|---|---|
| 6.1.4.1. Bakterien
Bakterien werden bei 37 °C im Schüttelinkubator (225 rpm; Novotron; Infors, Schweiz) oder im Brutschrank (Model B; Memmert, Deutschland) kultiviert. Bakterien können bei –80 °C eingefroren und so für Jahre gelagert werden. Dafür werden Kulturen bis zur Sättigung angezogen, Glycerin bis zu einer Konzentration von 20% (v/v) zugegeben und die Zellen in flüssigem Stickstoff eingefroren, bevor sie bei –80 °C gelagert werden. Eingefrorene Zellen können wieder in Kultur genommen werden, indem sie auf Eis aufgetaut werden und in Medium resuspendiert werden. LB-Medium: 10 g/l Pepton (GibcoBRL, USA) 5 g/l Hefeextrakt (GibcoBRL, USA) 10 g/l NaCl 15 g/l Agar (nur bei Festmedien)
5 g/l Hefeextrakt (GibcoBRL, USA) 0,5 g/l NaCl 4 g/l MgCl2 3,6 g/l Glucose 186 mg/l KCl . Nach dem Autoklavieren wird bei Selektivmedien noch 50 mg/ml Ampicillin zugegeben. 6.1.4.2. Hefen Hefezellen werden bei 30 °C im Schüttelinkubator (140 rpm; RFI-150; Infors, Schweiz) oder im Brutschrank (Heraeus, Deutschland) kultiviert. Hefen können bei –80 °C eingefroren und so für Jahre gelagert werden. Dafür werden Hefekulturen über Nacht angezogen, auf eine berechnete Zelldichte von OD600 50-100 eingestellt, Glycerin mit einer Konzentration von 15% (v/v) zugegeben und die Zellen in flüssigem Stickstoff eingefroren, bevor sie bei –80 °C gelagert werden. Eingefrorene Zellen können wieder in Kultur genommen werden, indem sie auf Eis aufgetaut werden und in Medium resuspendiert werden. |
2.1.5.1 Bakterien
Bakterien werden bei 37 °C im Schüttelinkubator (225 rpm; Novotron; Infors, Schweiz) oder im Brutschrank (Model B; Memmert, Deutschland) kultiviert. Bakterien können bei –80 °C eingefroren und so für Jahre gelagert werden. Dafür werden Kulturen bis zur Sättigung angezogen, Glycerin bis zu einer Konzentration von 20% (v/v) zugegeben und die Zellen in flüssigem Stickstoff eingefroren, bevor sie bei –80 °C gelagert werden. Eingefrorene Zellen können wieder in Kultur genommen werden, indem sie auf Eis aufgetaut werden und in Medium resuspendiert werden. LB-Medium: 10 g/l Pepton (GibcoBRL, USA)
[...] SOC-Medium: 20 g/l Pepton (GibcoBRL, USA)
[...] Nach dem Autoklavieren werden bei Selektivmedien noch entsprechende Antibiotika zugegeben: [...] 50 mg/ml Ampicillin [...] 2.1.5.2 Hefen Hefezellen werden bei 30 °C im Schüttelinkubator (140 rpm; RFI-150; Infors, Schweiz) oder im Brutschrank (Heraeus, Deutschland) kultiviert. Hefen können bei –80 °C eingefroren und so für Jahre gelagert werden. Dafür werden Hefekulturen über Nacht angezogen, auf eine berechnete Zelldichte von OD600 50-100 eingestellt, Glycerin mit einer Konzentration von 15% (v/v) zugegeben und die Zellen in flüssigem Stickstoff eingefroren, bevor sie bei –80 °C gelagert werden. Eingefrorene [Seite 33] Zellen können wieder in Kultur genommen werden, indem sie auf Eis aufgetaut werden und in Medium resuspendiert werden. |
Ein Verweis auf die Quelle fehlt. |
|
| [15.] Hw/Fragment 029 12 - Diskussion Bearbeitet: 31. August 2014, 12:34 Schumann Erstellt: 27. August 2014, 12:39 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 29, Zeilen: 12-16 |
Quelle: Blume 2003 Seite(n): 31, Zeilen: letzter Abschnitt |
|---|---|
| 6.1.4. Medien und Kultivierung der Zellen
Zum Ansetzen von Lösungen und Medien wird destilliertes entionisiertes Wasser verwendet. Stammlösungen und Flüssigmedien für die sterile Anzucht werden 20 min bei 200 kPa autoklaviert bzw. hitzelabile Lösungen sterilfiltriert (Satorius- Membranfilter, Porengröße 0,2μm). |
2.1.5 Medien und Kultivierung der Zellen
Zum Ansetzen von Lösungen und Medien wird destilliertes entionisiertes Wasser verwendet. Stammlösungen und Flüssigmedien für die sterile Anzucht werden 20 min bei 200 kPa autoklaviert bzw. hitzelabile Lösungen sterilfiltriert (Satorius- Membranfilter, Porengröße 0,2 μm). |
Ein Verweis auf die Quelle fehlt. |
|
| [16.] Hw/Fragment 020 01 - Diskussion Bearbeitet: 31. August 2014, 12:31 Schumann Erstellt: 27. August 2014, 13:01 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 20, Zeilen: 1-26 |
Quelle: Blume 2003 Seite(n): 78, 79, Zeilen: 78: 24ff - 79: 1ff |
|---|---|
| Der pYES2/lacZ-Vektor enthält das lacZ-Gen, das für die β-Galactosidase codiert, ein Enzym, das von verschiedenen Galactosederivaten hydrolytisch Galactose abspaltet. Die Aktivität dieses Enzyms kann in einem β-Gal-Assay nachgewiesen werden. In den mit dem pYES2/lacZ-Vektor transformierten Hefen konnte mit diesem Assay β-Galactosidase-Aktivität detektiert werden. Gleichzeitig wurden mittels Morgan- Elson-Test in den mit pYES2/2epi transformierten Hefen UDP-GlcNAc-2-Epimerase- Aktivitäten nachgewiesen. Damit läßt sich die UDP-GlcNAc-2-Epimerase/ManNAc-Kinase auch in S. cerevisiae als aktives Protein exprimieren. Um die maximale Proteinausbeute in Abhängigkeit von der Galactose-Induktion zu ermitteln, wurden zu verschiedenen Zeitpunkten Aliquots der Hefezellen geerntet und auf UDP-GlcNAc-2-Epimerase/ManNAc-Kinase bzw. β-Galactosidase untersucht. Werden jeweils die gemessenen Enzymaktivitäten auf die Proteinmengen bezogen, d.h. die spezifischen Enzymaktivitäten ermittelt, so läßt sich kein Zusammenhang zwischen der Galactose-Induktionszeit und der jeweiligen spezifischen Enzymaktivität erkennen. Nach einer sechsstündigen Galactose-Inkubation konnte eine geringfügig höhere Enzymkonzentration detektiert werden als zu den anderen Zeitpunkten. Aus den jeweiligen Aktivitätsbestimmungen konnte die Menge an überexprimiertem löslichem Protein pro Liter Hefekultur ermittelt werden. Da keine Relation zwischen der Zeit der Galactose-Induktion und der spezifischen Aktivität der Enzyme beobachtet wurde, wurde dieser Wert exemplarisch für t = 6 h ermittelt, wo die jeweiligen Kulturen eine OD600 von etwa 3 hatten. Für die β-Galactosidase lag dieser Wert bei etwa 90 μg/L Kultur und für die UDPGlcNAc- 2-Epimerase/ManNAc-Kinase bei etwa 34 μg/L Kultur. Die Firma Invitrogen gibt für die Expression von Proteinen in pYES-Systemen Werte von 0,1-100 mg/L an. Die Proteinexpression ist somit unter sub-optimalen Expressionsbedingungen durchgeführt worden, da die für die UDP-GlcNAc-2-Epimerase/ManNAc-Kinase erzielten Proteinmengen mit 34 μg/L weit unter den erwarteten Werten von mindestens 100 μg/L liegt und dieser Wert auch nicht mit der β-Galactosidase erzielt wurde. | Der pYES2/lacZ-Vektor enthält das lacZ-Gen, das für die β-Galactosidase codiert, ein Enzym, das von verschiedenen Galactosederivaten hydrolytisch Galactose abspaltet. Die Aktivität dieses Enzyms kann in einem β-Gal-Assay nachgewiesen werden. In den mit dem pYES2/lacZ-Vektor transformierten Hefen konnte mit diesem Assay β-Galactosidase-Aktivität detektiert werden. Gleichzeitig wurden mittels Morgan- Elson-Test in den mit pYES2/2epi transformierten Hefen UDP-GlcNAc-2-Epimerase- Aktivitäten nachgewiesen. Damit läßt sich die UDP-GlcNAc-2-Epimerase/ManNAc- Kinase auch in der Hefe S. cerevisiae als aktives Protein exprimieren.
Um die maximale Proteinausbeute in Abhängigkeit von der Galactose-Induktion zu ermitteln, wurden zu verschiedenen Zeitpunkten Aliquots der Hefezellen geerntet und auf UDP-GlcNAc-2-Epimerase/ManNAc-Kinase bzw. β-Galacotsidase untersucht. Werden jeweils die gemessenen Enzymaktivitäten auf die Proteinmengen bezogen, d.h. die spezifischen Enzymaktivitäten ermittelt, so läßt sich kein Zusammenhang zwischen der Galactose-Induktionszeit und der jeweiligen spezifischen Enzymaktivität erkennen. [Seite 79] Nach einer sechsstündigen Galactose-Inkubation konnte eine geringfügig höhere Enzymmenge detektiert werden als zu den anderen Zeitpunkten. Im SDS-Polyacrylamidgel konnte jedoch zu keinem Zeitpunkt β-Galactosidase bzw. UDP-GlcNAc-2-Epimerase/ManNAc-Kinase nachgewiesen werden. Aus den jeweiligen Aktivitätsbestimmungen konnte die Menge an überexprimiertem löslichem Protein pro Liter Hefekultur ermittelt werden. Da keine Relation zwischen der Zeit der Galactose-Induktion und der spezifischen Aktivität der Enzyme beobachtet wurde, wurde dieser Wert exemplarisch für t = 6 h ermittelt, wo die jeweiligen Kulturen eine OD600 von etwa 3 hatten. Für die β-Galactosidase lag dieser Wert bei etwa 90 μg/l Kultur und für die UDP-GlcNAc-2-Epimerase/ManNAc-Kinase bei etwa 34 μg/l Kultur. Die Firma Invitrogen gibt für die Expression von Proteinen in pYES-Systemen Werte von 0,1-100 mg/l an. Für das lacZ-Gen gibt es Angaben für den Genestorm- Kontrollvektor pYES/GS. In diesem Vektor liegt die Expressionsrate für die β-Galactosidase bei 10-15 μg/l/OD600. Die erzielte Expression von 30 μg/l/OD600 β-Galactosidase liegt damit etwa doppelt so hoch wie die Erwartungswerte. Die Proteinexpression ist somit unter optimalen Expressionsbedingungen durchgeführt worden; jedoch liegen die für die UDP-GlcNAc-2-Epimerase/ManNAc-Kinase erzielten Proteinmengen mit 34 μg/l weit unter den erwarteten Werten von mindestens 100 μg/l. |
Ein Verweis auf die Quelle fehlt. |
|
| [17.] Hw/Fragment 019 01 - Diskussion Bearbeitet: 31. August 2014, 12:30 Schumann Erstellt: 27. August 2014, 12:58 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, KomplettPlagiat, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 19, Zeilen: 1-13 |
Quelle: Blume 2003 Seite(n): 78, Zeilen: 8-23 |
|---|---|
| Anschließend wurde der pYES2/2epi-Vektor in den S. cerevisiae-Stamm INVSc1 transformiert. Als Kontrollen wurden zusätzlich der Leervektor und ein pYES2/lacZ-Konstrukt (Invitrogen), das zur Expression der β-Galactosidase führt, transformiert. Die Selektion der transgenen Hefezellen erfolgte durch die Abwesenheit von Uracil im Medium. Der Uracil-auxotrophe Hefestamm INVSc1 kann Uracil durch einen Defekt im URA3-Gen nicht mehr selbst synthetisieren. Dieser Defekt wird durch die Anwesenheit des pYES2-Vektors, der das URA3- Gen trägt, komplementiert. Die transgenen Hefen wurden in größeren Mengen in Uracildefizientem Medium angezogen. Bei einer OD600 von etwa 1,0 wurde mit 2% Galactose induziert, so daß das UDP-GlcNAc-2-Epimerase/ManNAc-Kinase-Gen abgelesen werden konnte. Zu den Zeitpunkten 0, 2, 4, 6, 8, 10 und 24 h nach der Galactose-Induktion wurde jeweils ein Aliquot der Hefezellen geerntet und mit Glasperlen mechanisch aufgeschlossen. Nach Zentrifugation wurde der proteinhaltige Überstand auf Enzymaktivitäten untersucht und die Anwesenheit der UDP-GlcNAc-2-Epimerase/ManNAc-Kinase bzw. der β-Galactosidase in [einem SDS-Polyacrylamidgel versucht nachzuweisen.] | Anschließend wurde der pYES2/2epi-Vektor in den S. cerevisiae Stamm INVSc1 transformiert. Als Kontrollen wurden zusätzlich der Leervektor und ein pYES2/lacZ-Konstrukt (Invitrogen), das zur Expression der β-Galactosidase führt, transformiert. Die Selektion der transgenen Hefezellen erfolgte durch die Abwesenheit von Uracil im Medium. Der Uracil-auxotrophe Hefestamm INVSc1 kann Uracil durch einen Defekt im URA3-Gen nicht mehr selbst synthetisieren. Dieser Defekt wird durch die Anwesenheit des pYES2- Vektors, der das URA3-Gen trägt, komplementiert.
Die transgenen Hefen wurden in größeren Mengen in uracildefizientem Medium angezogen. Bei einer OD600 von etwa 1,0 wurde mit 2% Galactose induziert, so daß das UDP-GlcNAc-2-Epimerase/ManNAc-Kinase-Gen abgelesen werden konnte. Zu den Zeitpunkten 0, 2, 4, 6, 8, 10 und 24 h nach der Galactose-Induktion wurde jeweils ein Aliquot der Hefezellen geerntet und mit Glasperlen mechanisch aufgeschlossen. Nach Zentrifugation wurde der proteinhaltige Überstand auf Enzymaktivitäten untersucht und die Anwesenheit der UDP-GlcNAc-2-Epimerase/ManNAc-Kinase bzw. der β-Galactosidase in einem SDS-Polyacrylamidgel versucht nachzuweisen. |
Ein Verweis auf die Quelle fehlt. |
|
| [18.] Hw/Fragment 016 01 - Diskussion Bearbeitet: 31. August 2014, 12:28 Schumann Erstellt: 27. August 2014, 13:57 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 16, Zeilen: 1ff (komplett) |
Quelle: Blume 2003 Seite(n): 108, Zeilen: 1ff |
|---|---|
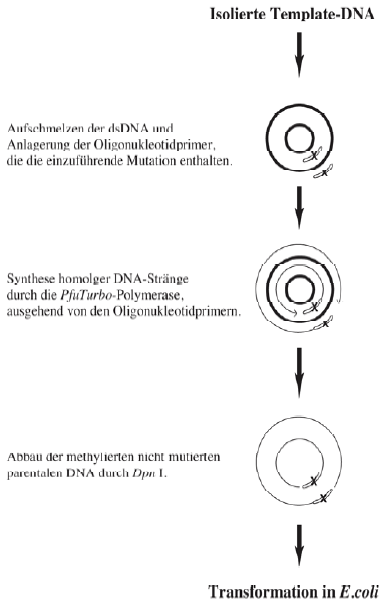
Abbildung 6: Prinzip der QuickChangeTM Site-Directed-Mutagenesis. Doppelsträngige Plasmid-DNA wird bei 95 °C denaturiert, so daß sich nach dem Auftrennen der Doppelstrang-DNA Oligonukleotidprimer, mit der einzuführenden Mutation, anlagern können. Ausgehend von diesen Primern werden von der PfuTurbo-DNA-Polymerase komplementäre Stränge synthetisiert. Anschließend wird die parentale, methylierte und nicht mutierte DNA selektiv durch die Endonuklease Dpn I abgebaut. Die circuläre, doppelsträngige DNA mit der eingeführten Mutation wird in kompetente E. coli-Zellen transformiert. |
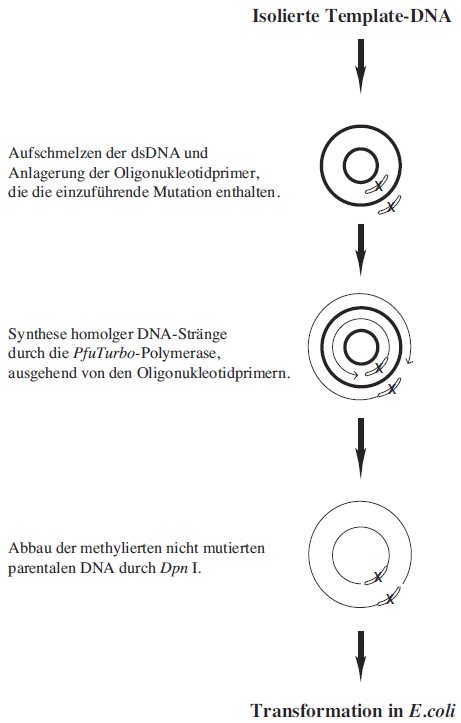
Abbildung 3.15: Prinzip der QuickChangeTM Site-Directed-Mutagenesis. Doppelsträngige Plasmid-DNA wird bei 95 °C denaturiert, so daß sich Oligonukleotidprimer, die die einzuführende Mutation enthalten, anlagern können. Ausgehend von diesen Primern werden von der PfuTurbo-DNA-Polymerase komplementäre Stränge synthetisiert. Anschließend wird die parentale, methylierte und nicht mutierte DNA selektiv durch die Endonuklease Dpn I abgebaut. Die circuläre, doppelsträngige DNA mit der eingeführten Mutation wird in kompetente E. coli-Zellen transformiert. |
Ein Verweis auf die Quelle fehlt. |
|
| [19.] Hw/Fragment 011 10 - Diskussion Bearbeitet: 31. August 2014, 12:27 Schumann Erstellt: 27. August 2014, 17:07 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 11, Zeilen: 10-23 |
Quelle: Blume 2003 Seite(n): 25, 26, Zeilen: 25: 33ff - 26: 1ff |
|---|---|
| Durch Arbeiten an hämatopoetischen Zellinien, welche keine Expression der UDP-GlcNAc-2- Epimerase mehr aufwiesen, konnte die zentrale Rolle des Enzyms für die Regulation der Sialierung von Glycoproteinen und Glycolipiden der Plasmamembran gezeigt werden (Keppler et al., 1999). Solche Zellen sind nicht mehr in der Lage, eigenständig Sialinsäuren zu bilden und weisen zahlreiche funktionelle Defekte auf, wie etwa das Fehlen der homophilen Interaktion von CD22 oder von P-Selektin mit seinen Liganden (Keppler et al., 1999). Wird die UDP-2- GlcNAc-Epimerase/ManNAc-Kinase durch gezielte Mutagenese in der Maus ausgeschaltet, sterben die Embryonen spätestens am Tag 8,5 der Embryonalentwicklung (Schwarzkopf et al., 2002). Diese Ergebnisse beweisen die essentielle Rolle der Sialinsäuren für die Embryonalentwicklung,
Der Reaktionsmechanismus der UDP-GlcNAc-2-Epimerase ist relativ gut untersucht. Sie benötigt im Gegensatz zu anderen Epimerasen (z. B. die UDP-GlcNAc-4-Epimerase) kein Coenzym, wie etwa NADH, für die katalytische Reaktion, welche höchstwahrscheinlich in drei Schritten erfolgt (Tanner 2002). |
Die zentrale Rolle der UDP-GlcNAc-2-Epimerase/ManNAc-Kinase für die Regulation der Sialylierung von Glycoproteinen und Glycolipiden der Plasmamembran konnte durch Arbeiten an hämatopoietischen Zellinien gezeigt werden, die keine Expression des Enzyms mehr aufwiesen (Keppler et al., 1999). Solche Zellen sind nicht mehr in der Lage, eigenständig Sialinsäuren zu bilden und weisen zahlreiche funktionelle Defekte auf, so etwa die fehlende homophile Interaktion des Siglec2, die Interaktion des
[Seite 26] P-Selektins mit seinen Liganden (Keppler et al., 1999) oder auch die Reduktion der Zell-Matrix-Interaktion (Suzuki et al., 2002). Wird die UDP-GlcNAc-2-Epimerase/ ManNAc-Kinase durch gezielte Mutagenese in der Maus ausgeschaltet, sterben die Embryonen spätestens am Tag 8,5 der Embryonalentwicklung (Schwarzkopf et al., 2002). Diese Ergebnisse zeigen, daß die Sialinsäuren für die Embryonalentwicklung essentiell sind. [...] [...] Der Reaktionsmechanismus der UDP-GlcNAc-2-Epimerase ist relativ gut untersucht (Abb. 1.5). Im Unterschied zu anderen Epimerasen, etwa der UDP-GalNAc-4- Epimerase, benötigt die UDP-GlcNAc-2-Epimerase für die katalytische Reaktion kein Coenzym, wie z.B. NAD+. Die Reaktion erfolgt höchstwahrscheinlich in mehreren Schritten (Tanner, 2002). |
Ein Verweis auf die Quelle fehlt. |
|
| [20.] Hw/Fragment 010 21 - Diskussion Bearbeitet: 31. August 2014, 12:25 Schumann Erstellt: 27. August 2014, 17:14 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 10, Zeilen: 21-29 |
Quelle: Blume 2003 Seite(n): 26, Zeilen: 7-16 |
|---|---|
| Bisher wurde die UDP-GlcNAc-2-Epimerase/ManNAc-Kinase aus Ratte (Stäsche et al., 1997), Maus (Horstkorte et al., 1999) und Mensch (Lucka et al., 1999) kloniert. Es zeigte sich, daß die Homologie der Aminosäuresequenz sehr hoch ist: von 722 Aminosäuren sind zwischen Ratte und Maus vier, zwischen Ratte und Mensch zehn und zwischen Maus und Mensch zwölf unterschiedlich. Punktmutationen konservierter Aminosäuren führen zu einem selektiven Verlust der Enzymaktivität der jeweils betroffenen Domäne, ohne die Aktivität der anderen zu beeinflussen (Effertz et al., 1999). Sequenzvergleiche mit Zuckerkinasen beziehungsweise bakteriellen UDP-GlcNAc-2-Epimerasen legen zwei funktionelle Domänen nahe, eine N-terminale Epimerase- und eine C-terminale Kinase-Domäne. | Die UDP-GlcNAc-2-Epimerase/ManNAc-Kinase wurde bisher aus Ratte (Stäsche et al., 1997), Maus (Horstkorte et al., 1999) und Mensch (Lucka et al., 1999) kloniert. Die Homologie der Aminosäuresequenz ist sehr hoch, zwischen Ratte und Maus sind 4, zwischen Ratte und Mensch 10 und zwischen Maus und Mensch 12 von 722 Aminosäuren unterschiedlich. Sequenzvergleiche mit Zuckerkinasen bzw. bakteriellen UDP-GlcNAc-2-Epimerasen legen zwei funktionelle Domänen nahe, eine N-terminale Epimerase- und eine C-terminale Kinasedomäne. Punktmutationen konservierter Aminosäuren führen zu einem selektiven Verlust der Enzymaktivität der jeweils betroffenen Domäne, ohne die Aktivität der anderen Domäne zu beeinflussen (Effertz et al., 1999). |
Ein Verweis auf die Quelle fehlt. Im nächsten Satz wird auf Blume et al. (2004) verwiesen, eine Publikation, die Überschneidungen mit der Quelle haben mag, aber auf Englisch verfasst ist. |
|
| [21.] Hw/Fragment 008 01 - Diskussion Bearbeitet: 31. August 2014, 12:24 Schumann Erstellt: 27. August 2014, 17:22 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 8, Zeilen: 1-13 |
Quelle: Blume 2003 Seite(n): 18, Zeilen: 6ff |
|---|---|
| [Ebenso ist die] Asialoform des Nukleoporins p62, welches den aktiven Proteintransport vom Cytosol in den Zellkern unterstützt, in seiner Aktivität stark reduziert (Emig et al., 1995). Für Erythropoetin wurden ähnliche Beobachtungen gemacht (Wasley et al., 1991); dies ist allerdings auf die stark verminderte Halbwertzeit des Asialoproteins im Blut zurückzuführen (Egrie und Brown, 2001). Die Zirkulationszeit von Blutzellen wird ebenfalls durch ihren Gehalt an terminalen Sialinsäuren reguliert: Mit der Zeit verlieren Erythrozyten und Thrombozyten Sialinsäuren oder Sialoglycoproteine und werden dann von Zellen des reticuloendothelialen Systems erkannt und abgebaut (Kluge et al., 1992; Schlepper-Schäfer et al., 1980). Des Weiteren erkennen in der Leber lokalisierte Asialoglycoproteinrezeptoren Serumglycoproteine und Antigen- Antikörperkomplexe nach Verlust ihrer Sialinsäuren, so daß sie endocytiert und in den Lysosomen abgebaut werden können (Ashwell und Harford, 1982). Ob die Zellen beziehungsweise Glycoproteine ihre Sialinsäuren durch nicht-enzymatische Hydrolyse oder durch Sialidasen verlieren, wurde bis heute nicht geklärt. | Die Asialoform des Nukleoporins p62, das den aktiven Proteintransport vom Cytosol in den Zellkern unterstützt, zeigt eine stark reduzierte Aktivität (Emig et al., 1995). Eine ähnliche Beobachtung wurde für Erythropoietin gemacht (Wasley et al., 1991), ein Hormon, das die Bildung von Erythrocyten stimuliert. Hierbei beruht die verringerte biologische Aktivität des Asialoproteins allerdings auf einer reduzierten Halbwertszeit im Blut (Egrie und Brown, 2001). [...]. Die Zirkulationszeit von Blutzellen wird ebenfalls durch ihren Gehalt an terminalen Sialinsäuren reguliert. Erythrozyten und Thrombozyten verlieren bei ihrer Alterung ihre Sialinsäuren bzw. Salinsäure-haltige Strukturen und werden dann von Makrophagen erkannt und phagocytiert (Schlepper-Schäfer et al., 1980; Kluge et al., 1992). Ähnliches wird bei Serumglycoproteinen und Antigen-Antikörperkomplexen beobachtet. Sie werden nach Verlust ihrer terminalen Sialinsäuren durch den Asialoglycoproteinrezeptor der Leber erkannt, endocytiert und abgebaut (Ashwell und Harford, 1982). Auf welche Weise die Zellen bzw. Glycoproteine ihre Sialinsäuren bzw. Sialinsäure-haltigen Strukuren verlieren, ist bis heute nicht geklärt. |
Ein Verweis auf die Quelle fehlt. Die Übernahme beginnt auf der Vorseite: Fragment 007 32. |
|
| [22.] Hw/Fragment 007 32 - Diskussion Bearbeitet: 31. August 2014, 12:22 Schumann Erstellt: 27. August 2014, 20:04 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 7, Zeilen: 32-34 |
Quelle: Blume 2003 Seite(n): 18, Zeilen: 3-6 |
|---|---|
| Für die biologische Funktion einiger Glycoproteine ist die Präsenz von Sialinsäuren unabdingbar. So führt zum Beispiel die Desialylierung des Somatostatinrezeptors zu einer wesentlich schlechteren Ligandenbindung (Rens-Domiano und Reisine, 1991). | Die Präsenz von Sialinsäuren ist wichtig für die biologische Funktion einiger Glycoproteine. So führt z.B. die Desialylierung des Somatostatinrezeptors zu einer Konformationsänderung und damit zu einer deutlich schlechteren Ligandenbindung (Rens-Domiano und Reisine, 1991). |
Ein Verweis auf die Quelle fehlt. Fortsetzung auf der nächsten Seite: Fragment 008 01. |
|
| [23.] Hw/Fragment 007 01 - Diskussion Bearbeitet: 31. August 2014, 12:20 Schumann Erstellt: 27. August 2014, 19:58 (Hindemith) | Blume 2003, Fragment, Gesichtet, Hw, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 7, Zeilen: 1-5 |
Quelle: Blume 2003 Seite(n): 16, Zeilen: 31-36 |
|---|---|
| [Seine Spezifität für bestimmte Sialinsäuretypen ist streng abhängig von der] Sialylierung der Wirtszelle, z.B. von Mensch, Huhn oder Schwein (Suzuki et al., 2000). So kann es durch Kreuzinfektionen von Influenza A-Viren, insbesondere bei Haustieren untereinander und mit Menschen, verstärkt durch horizontalen Gentransfer unter den Virusspezies vorkommen, so daß sich tierische Viren an humane Zellen anpassen und so zu saisonalen Epidemien führen (Ito et al., 1998). | Seine Spezifität für bestimmte Sialinsäuretypen hängt streng von der Sialylierung der Wirtszelle (z.B. von Mensch, Huhn oder Schwein) ab (Suzuki et al., 2000). Durch Kreuzinfektionen von Influenza A-Viren, insbesondere bei Haustieren untereinander und mit Menschen, verstärkt durch horizontalen Gentransfer unter den Virenspezies, können sich tierische Viren an die Sialylierung humaner Zellen anpassen und so zu saisonalen Epidemien führen (Ito et al., 1998). |
Ein Verweis auf die Quelle fehlt. |
|