88 gesichtete, geschützte Fragmente: Plagiat
| [1.] Mpl/Fragment 031 01 - Diskussion Bearbeitet: 27. November 2014, 15:14 PlagProf:-) Erstellt: 8. November 2014, 11:35 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 31, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 33, Zeilen: 1 ss. |
|---|---|
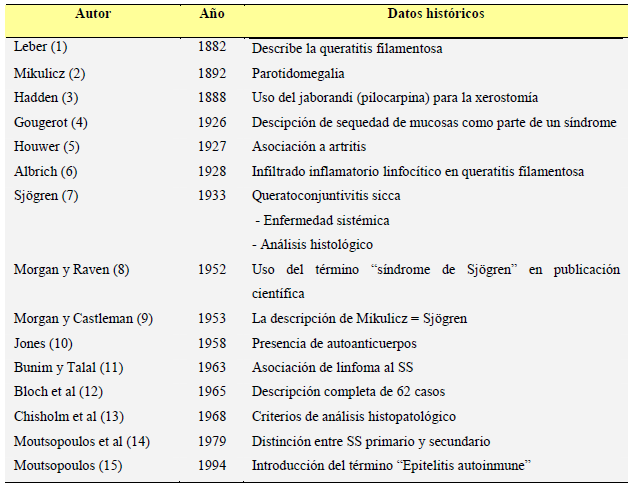
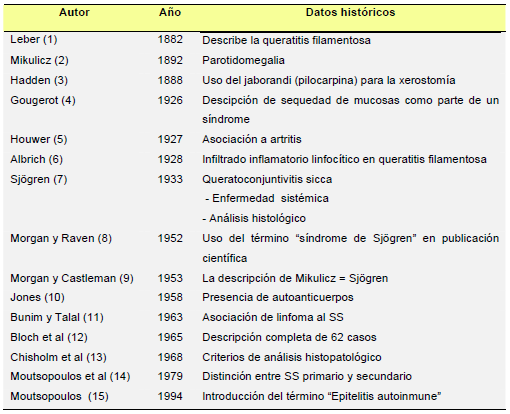
| 1. SÍNDROME DE SJÖGREN PRIMARIO: GENERALIDADES
El síndrome de Sjögren (SS) es una enfermedad autoinmune sistémica que se caracteriza fundamentalmente por la presencia de sequedad ocular (xeroftalmía) y bucal (xerostomía), debido a la infiltración de las glándulas lagrimales y salivares por células linfoplasmocitarias. Estos infiltrados originan una destrucción progresiva de las glándulas exocrinas, con la consiguiente disminución de las secreciones glandulares y la aparición de sintomatología relacionada con la sequedad de las mucosas infiltradas. La hiperactividad de los linfocitos B periféricos es el principal dato inmunológico presente en el SS. Históricamente (Tabla 1) (1-15), las primeras descripciones de pacientes con sequedad de mucosas se realizaron a finales del siglo pasado, aunque no fue hasta 1933 cuando un oftalmólogo sueco, Henrik Sjögren (Figura 1), englobó dichas manifestaciones en un trastorno autoinmune generalizado que presentaba además otros signos de afección sistémica, como artritis o anemia (7).
FIGURA 1. Henrik Sjögren |
1. SÍNDROME DE SJÖGREN PRIMARIO: GENERALIDADES
El síndrome de Sjögren (SS) es una enfermedad autoinmune sistémica que se caracteriza fundamentalmente por la presencia de sequedad ocular (xeroftalmía) y bucal (xerostomía), debido a la infiltración de las glándulas lagrimales y salivares por células linfoplasmocitarias. Estos infiltrados originan una destrucción progresiva de las glándulas exocrinas, con la consiguiente disminución de las secreciones glandulares y la aparición de sintomatología relacionada con la sequedad de las mucosas infiltradas. La hiperactividad de los linfocitos B periféricos es el principal dato inmunológico presente en el SS. Históricamente (Tabla 1) (1-15), las primeras descripciones de pacientes con sequedad de mucosas se realizaron a finales del siglo pasado, aunque no fue hasta 1933 cuando un oftalmólogo sueco, Henrik Sjögren (Figura 1), englobó dichas manifestaciones en un trastorno autoinmune generalizado que presentaba además otros signos de afección sistémica, como artritis o anemia (7).
FIGURA 1. Henrik Sjögren |


Brito Zerón (2006) no se menciona. Nota que las referencias a la literatura son iguales en ambos textos (y no se han documentado). La figura se ha cambiado, pero sin dar alguna referencia, un posible orígen es [1]. |
|
| [2.] Mpl/Fragment 032 01 - Diskussion Bearbeitet: 27. November 2014, 15:18 PlagProf:-) Erstellt: 8. November 2014, 13:11 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 32, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 34, 35, Zeilen: 34: 1 ss. ; 35: 1-2 |
|---|---|
| TABLA 1. Fechas a destacar en la historia del SS
1.1. Epidemiología El SS es probablemente la enfermedad autoinmune más frecuente en nuestro medio aunque su habitual pobreza en sus manifestaciones clínicas, especialmente en estadios evolutivos tempranos, conlleva que esté a menudo infradiagnosticada. Afecta predominantemente al sexo femenino, con una relación respecto al varón de 9-10:1. Los estudios realizados en varones no han mostrado diferencias significativas en las manifestaciones clínicas respecto a las que presentan las mujeres, aunque sí se observa una tendencia a la negatividad de los marcadores inmunológicos. En la mayoría de los casos, el SS aparece entre los 40 y los 60 años, aunque también se han descrito casos en edades más tempranas de la vida, y casos en edad geriátrica. |
TABLA 1. Fechas a destacar en la historia del SS
1.1. Epidemiología El SS es probablemente la enfermedad autoinmune más frecuente en nuestro medio aunque su habitual pobreza en sus manifestaciones clínicas, especialmente en estadios evolutivos tempranos, conlleva que esté a menudo infradiagnosticada. Afecta predominantemente al sexo femenino, con una relación respecto al varón de 9-10:1. Los estudios realizados en varones no han mostrado diferencias significativas en las manifestaciones clínicas respecto a las que presentan las mujeres, aunque sí se observa una tendencia a la negatividad de los marcadores inmunológicos. En la mayoría de los casos, el SS aparece entre [página 35] los 40 y los 60 años, aunque también se han descrito casos en edades más tempranas de la vida, y casos en edad geriátrica. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). Donde hay un salto de página en la fuente hay un aparte en la tesis de M. P. |
|
| [3.] Mpl/Fragment 033 01 - Diskussion Bearbeitet: 27. November 2014, 15:23 PlagProf:-) Erstellt: 8. November 2014, 13:19 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 33, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 35, 36, Zeilen: 35: 1 ss.; 36: 1ff |
|---|---|
| Los estudios realizados en otras enfermedades autoinmunes sistémicas muestran una prevalencia de SS en el 7% de pacientes con artritis reumatoide (AR) (16), en el 20-30% de los pacientes con esclerosis sistémica (ES) (17) o lupus eritematoso sistémico (LES) (18,19). Se ha descrito síndrome seco en el 42% de 55 pacientes con enfermedad mixta del tejido conectivo (EMTC) (20). La relación con otras enfermedades autoinmunes, como la esclerosis múltiple (EM) (21,22) o la enfermedad celiaca (23), también ha sido analizada recientemente.
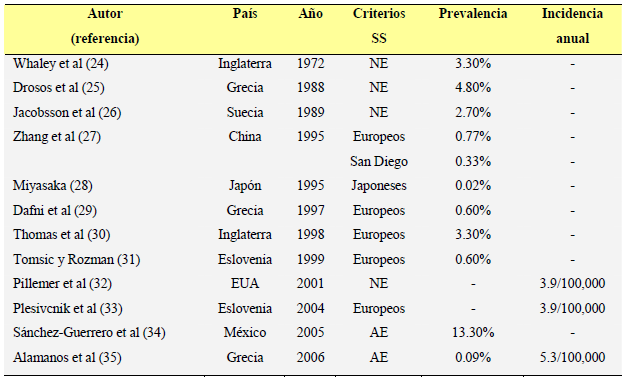
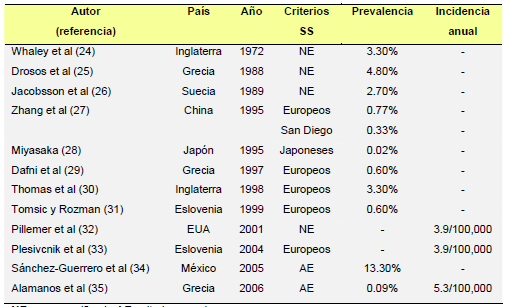
La prevalencia del SSp varía del 0.02 al 13.3% de acuerdo a los estudios publicados (24- 35) (Tabla 2). Sin embargo, los distintos criterios clasificatorios y los métodos epidemiológicos utilizados dificultan la interpretación de estas diferencias. En los estudios muestrales, la prevalencia del SSp varía del 0.77 al 3.3%. Se han encontrado prevalencias similares en los estudios epidemiológicos poblacionales, (0.02 al 2.70%) (26-28). Sin embargo, se han publicado prevalencias superiores (3.3-4.8%) (24,25) en ciertos grupos poblacionales como en población geriátrica hospitalizada y, recientemente, Sanchez- Guerrero et al (34), encontraron una prevalencia del 13.3% en 300 pacientes ambulatorios pertenecientes a los servicios de medicina interna y reumatología en un hospital mexicano de tercer nivel. La incidencia del SSp en la población general se ha analizado en 3 estudios, y a diferencia de la variabilidad encontrada en los estudios de prevalencia del SSp, los estudios de incidencia han encontrado resultados muy similares. Pillemer et al (en población estadounidense –32-) y Plesivcnik et al (en población eslovena –33-) encontraron una incidencia de 3,9 por 100,000 habitantes, mientras que Alamanos et al (35), encontraron una incidencia de 5,3 por 100,000 habitantes en población griega. |
Los estudios realizados en otras enfermedades autoinmunes sistémicas muestran una prevalencia de SS en el 7% de pacientes con artritis reumatoide (AR) (16), en el 20-30% de los pacientes con esclerosis sistémica (ES) (17) o lupus eritematoso sistémico (LES) (18,19). Se ha descrito síndrome seco en el 42% de 55 pacientes con enfermedad mixta del tejido conectivo (EMTC) (20). La relación con otras enfermedades autoinmunes, como la esclerosis múltiple (EM) (21,22) o la enfermedad celiaca (23), también ha sido analizada recientemente.
La prevalencia del SSp varía del 0.02 al 13.3% de acuerdo a los estudios publicados (24-35) (Tabla 2). Sin embargo, los distintos criterios clasificatorios y los métodos epidemiológicos utilizados dificultan la interpretación de estas diferencias. En los estudios muestrales, la prevalencia del SSp varía del 0.77 al 3.3%. Se han encontrado prevalencias similares en los estudios epidemiológicos poblacionales, (0.02 al 2.70%) (26-28). Sin embargo, se han publicado prevalencias superiores (3.3-4.8%) (24,25) en ciertos grupos poblacionales como en población geriátrica hospitalizada y, recientemente, Sanchez-Guerrero et al (34), encontraron una prevalencia del 13.3% en 300 pacientes ambulatorios pertenecientes a los servicios de medicina interna y reumatología en un hospital mexicano de tercer nivel. La incidencia del SSp en la población general se ha analizado en 3 estudios, y a diferencia de la variabilidad encontrada en los estudios de prevalencia del SSp, los estudios de incidencia han encontrado resultados muy similares. Pillemer et al (en población estadounidense –32-) y Plesivcnik et al (en población eslovena – 33-) encontraron una incidencia de 3,9 por 100,000 habitantes, mientras que [página 36] Alamanos et al (35), encontraron una incidencia de 5,3 por 100,000 habitantes en población griega. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [4.] Mpl/Fragment 034 01 - Diskussion Bearbeitet: 27. November 2014, 15:27 PlagProf:-) Erstellt: 8. November 2014, 13:40 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 34, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 36, 37, Zeilen: 36: 3 ss.; 37: 1 ss. |
|---|---|
| TABLA 2. Prevalencia e incidencia del SSp
NE, no especificado;AE, criterios americano-europeos 1.2. Etiopatogenia No se conoce adecuadamente la etiopatogenia del SS, aunque se ha propuesto la existencia de factores genéticos predisponentes sobre los que podrían actuar factores exógenos (principalmente infecciones víricas) y factores neurohormonales. Las principales características patogénicas del SS son la infiltración glandular exocrina por linfocitos T y la hiperestimulación de los linfocitos B (Figura 2). El proceso autoinmune se inicia en el tejido glandular exocrino (epitelitis autoinmune), y podría estar desencadenado por agentes externos (principalmente virus con especial tropismo por el tejido glandular), bien directamente o bien a través de una reacción cruzada con moléculas propias (mimetismo molecular). Se produciría entonces una infiltración inflamatoria constituida en su mayor parte por linfocitos T CD4+, con la producción local de citocinas de predominio Th1 que contribuiría a perpetuar la respuesta inflamatoria en las glándulas salivales (36). |
TABLA 2. Prevalencia e incidencia del SSp
NE, no especificado;AE, criterios americano-europeos 1.2. Etiopatogenia No se conoce adecuadamente la etiopatogenia del SS, aunque se ha propuesto la existencia de factores genéticos predisponentes sobre los que podrían actuar factores exógenos (principalmente infecciones víricas) y factores neurohormonales. Las principales características patogénicas del SS son la infiltración glandular exocrina por linfocitos T y la hiperestimulación de los linfocitos B (Figura 2). El proceso autoinmune se inicia en el tejido glandular exocrino (epitelitis autoinmune), y podría estar desencadenado por agentes externos (principalmente virus con especial tropismo por el tejido glandular), bien directamente o bien a través de una reacción cruzada con moléculas propias (mimetismo molecular). Se produciría entonces una infiltración inflamatoria [página 37] constituida en su mayor parte por linfocitos T CD4+, con la producción local de citocinas de predominio Th1 que contribuiría a perpetuar la respuesta inflamatoria en las glándulas salivales (36). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [5.] Mpl/Fragment 035 01 - Diskussion Bearbeitet: 27. November 2014, 15:31 PlagProf:-) Erstellt: 8. November 2014, 13:50 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 35, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 37, Zeilen: 3 ss. |
|---|---|
| [La] hiperestimulación de linfocitos B por parte de los linfocitos T activados originaría la producción de numerosos autoanticuerpos.
FIGURA 2. Relaciones anatómicas de glándulas salivales y de sus conductos excretores
A) Diagrama de una glándula salival normal con sus conductos y acinos B) Infiltrado linfocitario que destruye los acinos y debilita el conducto
|
La hiperestimulación de linfocitos B por parte de los linfocitos T activados originaría la producción de numerosos autoanticuerpos.
FIGURA 2. Relaciones anatómicas de glándulas salivales y de sus conductos excretores
A) Diagrama de una glándula salival normal con sus conductos y acinos B) Infiltrado linfocitario que destruye los acinos y debilita el conducto
|
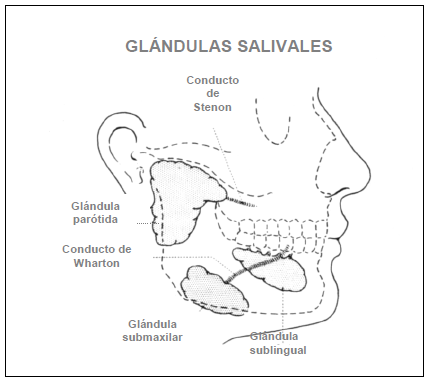
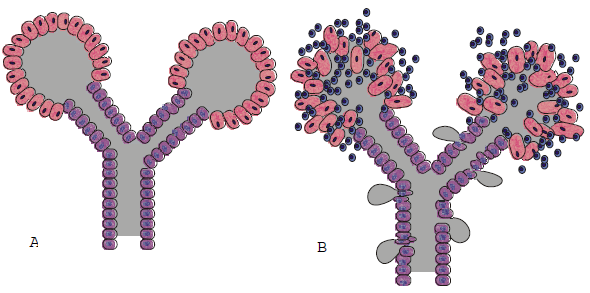
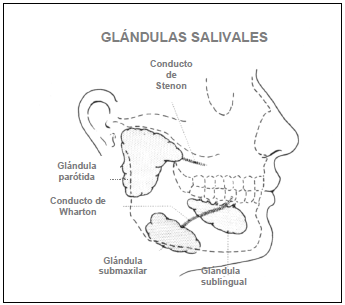
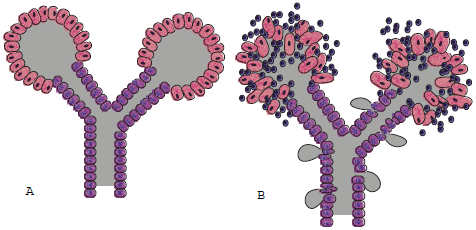
Brito Zerón (2006) no se menciona. |
|
| [6.] Mpl/Fragment 036 01 - Diskussion Bearbeitet: 27. November 2014, 16:08 Hindemith Erstellt: 8. November 2014, 13:56 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 36, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 38, 39, Zeilen: 38: 1 ss.; 39: 1-3 |
|---|---|
| 1.2.1. Autoantígenos
Se han sugerido recientemente moléculas propias como la α-fodrina y las aquoporinas como posibles autoantígenos. La α-fodrina forma parte del componente citoesquelético de diversas células eucariotas y está compuesta por dos cadenas heterodiméricas que se unen a la actina. Estudios recientes han detectado anticuerpos circulantes antifodrina en pacientes con SSp (37), especialmente en formas infantiles y juveniles (38,39). Las aquoporinas (AQP) son una familia de canales proteínicos de membrana cuya función es el transporte de agua. La AQP5 fue identificada en las glándulas submandibulares de rata, y está presente en las glándulas lagrimales y pulmón, indicando su función en la generación de saliva, lágrima y secreciones pulmonares (40). Se ha demostrado una distribución anormal de la AQP5 en las células acinares de pacientes con SS, respecto a otras entidades como sialoadenitis no específica, sarcoidosis, y ojo seco no autoinmune, en los que la AQP5 tiene una distribución similar a la población control. Al parecer existiría un defecto en el tráfico de la proteína, mas que en su síntesis, puesto que la cuantificación de AQP5 en la glándula lagrimal es similar en SS y controles (41-43). 1.2.2. Predisposición genética En familias con miembros afectos de SS se diagnostican otros casos de SS con mayor frecuencia que en la población general, y también existe una mayor incidencia de otras enfermedades autoinmunes y de autoanticuerpos en suero. La predisposición genética para el SSp podría estar ligada a los antígenos del complejo mayor de histocompatibilidad. Las frecuencias de los haplotipos B8, DR3, DR2 y especialmente del DRw 52, DQA1 0501 y DQB1 0201 son superiores en los pacientes con SS respecto a la población general. Las recientes técnicas de análisis multigénicos intentan identificar grupos de genes potencialmente implicados en la aparición del SS (44). |
1.2.1. Autoantígenos
Se han sugerido recientemente moléculas propias como la α-fodrina y las aquoporinas como posibles autoantígenos. La α-fodrina forma parte del componente citoesquelético de diversas células eucariotas y está compuesta por dos cadenas heterodiméricas que se unen a la actina. Estudios recientes han detectado anticuerpos circulantes antifodrina en pacientes con SSp (37), especialmente en formas infantiles y juveniles (38,39). Las aquoporinas (AQP) son una familia de canales proteínicos de membrana cuya función es el transporte de agua. La AQP5 fue identificada en las glándulas submandibulares de rata, y está presente en las glándulas lagrimales y pulmón, indicando su función en la generación de saliva, lágrima y secreciones pulmonares (40). Se ha demostrado una distribución anormal de la AQP5 en las células acinares de pacientes con SS, respecto a otras entidades como sialoadenitis no específica, sarcoidosis, y ojo seco no autoinmune, en los que la AQP5 tiene una distribución similar a la población control. Al parecer existiría un defecto en el tráfico de la proteína, mas que en su síntesis, puesto que la cuantificación de AQP5 en la glándula lagrimal es similar en SS y controles (41-43). 1.2.2. Predisposición genética En familias con miembros afectos de SS se diagnostican otros casos de SS con mayor frecuencia que en la población general, y también existe una mayor incidencia de otras enfermedades autoinmunes y de autoanticuerpos en suero. La predisposición genética para el SSp podría estar ligada a los antígenos del complejo mayor de histocompatibilidad. Las frecuencias de los haplotipos B8, DR3, DR2 y especialmente del DRw 52, DQA1 0501 y DQB1 0201 son [página 39] superiores en los pacientes con SS respecto a la población general. Las recientes técnicas de análisis multigénicos intentan identificar grupos de genes potencialmente implicados en la aparición del SS (44). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [7.] Mpl/Fragment 037 01 - Diskussion Bearbeitet: 27. November 2014, 17:20 PlagProf:-) Erstellt: 8. November 2014, 14:23 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 37, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 33, Zeilen: 1 ss. |
|---|---|
| [Los estudios monogénicos más] recientes se han centrado en el estudio de los polimorfismos de genes que codifican ciertas citocinas como la IL-10 (45-47), proteínas reguladoras como la proteína secretora rica en cisteína 3 (CRISP-3) (48) o la lectina fijadora de manano (MBL) (49).
1.2.3. Infecciones víricas Numerosos estudios sugieren que las infecciones víricas juegan un papel importante en la etiopatogenia del SS, especialmente en el caso de aquellos virus con un marcado tropismo salival. La orofaringe podría ser el reservorio de dichos virus, que permanecen habitualmente en estado latente bajo el control de la inmunidad local. En determinados individuos, genéticamente predispuestos, los virus podrían ser capaces de infectar las células epiteliales, e inducir la presentación de neoantígenos que iniciarían una respuesta anómala autoinmunitaria. Los principales agentes víricos implicados son los herpesvirus [virus de Epstein-Barr (VEB), citomegalovirus (CMV), virus herpes humano tipo 6 (VHH-6)], retrovirus [virus de la inmunodeficiencia humana (VIH) y virus de la leucemia T humana (HTLV-I), virus de la hepatitis (VHC) y parvovirus B19 (PV-B19). Mención especial merece el VHC, que podría ser el principal factor etiopatogénico en un subgrupo de pacientes con SS que presentan afección hepática y/o crioglobulinemia mixta (CM) (50). 1.3. Manifestaciones clínicas Si bien la sequedad ocular y bucal son los síntomas más frecuentes en el SS, las manifestaciones clínicas que pueden aparecer en el curso evolutivo de este síndrome son múltiples. En la gran mayoría de casos la infiltración linfocitaria queda confinada al tejido glandular salival y lagrimal, pero en ocasiones puede extenderse a localizaciones extraglandulares. |
Los estudios monogénicos más recientes se han centrado en el estudio de los polimorfismos de genes que codifican ciertas citocinas como la IL-10 (45-47), proteínas reguladoras como la proteína secretora rica en cisterna [sic] 3 (CRISP-3) (48) o la lectina fijadora de manano (MBL) (49).
1.2.3. Infecciones víricas Numerosos estudios sugieren que las infecciones víricas juegan un papel importante en la etiopatogenia del SS, especialmente en el caso de aquellos virus con un marcado tropismo salival. La orofaringe podría ser el reservorio de dichos virus, que permanecen habitualmente en estado latente bajo el control de la inmunidad local. En determinados individuos, genéticamente predispuestos, los virus podrían ser capaces de infectar las células epiteliales, e inducir la presentación de neoantígenos que iniciarían una respuesta anómala autoinmunitaria. Los principales agentes víricos implicados son los herpesvirus [virus de Epstein-Barr (VEB), citomegalovirus (CMV), virus herpes humano tipo 6 (VHH-6)], retrovirus [virus de la inmunodeficiencia humana (VIH) y virus de la leucemia T humana (HTLV-I), virus de la hepatitis (VHC) y parvovirus B19 (PV-B19). Mención especial merece el VHC, que podría ser el principal factor etiopatogénico en un subgrupo de pacientes con SS que presentan afección hepática y/o crioglobulinemia mixta (CM) (50). [página 40] 1.3. Manifestaciones clínicas Si bien la sequedad ocular y bucal son los síntomas más frecuentes en el SS, las manifestaciones clínicas que pueden aparecer en el curso evolutivo de este síndrome son múltiples. En la gran mayoría de casos la infiltración linfocitaria queda confinada al tejido glandular salival y lagrimal, pero en ocasiones puede extenderse a localizaciones extraglandulares. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [8.] Mpl/Fragment 038 01 - Diskussion Bearbeitet: 27. November 2014, 17:26 PlagProf:-) Erstellt: 8. November 2014, 19:31 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 38, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 40, Zeilen: 40: 7 ss. ; 41: 1 ss. |
|---|---|
| 1.3.1. Mucosa oral
Los síntomas relacionados con la xerostomía suelen iniciarse de forma insidiosa. Según el grado de intensidad de afección, el paciente puede presentar dificultad para hablar y comer (especialmente alimentos sólidos) y como consecuencia, perder peso, presentar halitosis, alteración del sabor de los alimentos, disestesias, sensación de ardor o quemazón bucal y labial e intolerancia para alimentos ácidos. En casos graves, el paciente suele llevar consigo una botella de agua para conseguir un alivio rápido de los síntomas. Con la hiposecreción salival se pierde el efecto tampón de la saliva y se incrementa la aparición de caries por el predominio de la flora favorecedora de la lesión dental (Streptococcus mutans), que puede agravarse con la presencia de enfermedad periodontal asociada. La hiposecreción salival conduce también al aumento de infecciones bucales, especialmente por Candida albicans (51). En los pacientes con SS, la prevalencia de candidiasis oral alcanza el 70%. Se manifiesta con síntomas de ardor y quemazón bucal, además de intolerancia a los alimentos ácidos y picantes. 1.3.2. Mucosa ocular Aparece en más del 90% de los pacientes y es la manifestación clínica más frecuente del SS. La expresión que refieren es la sensación constante de tener arena o tierra en lo ojos. Las molestias son más intensas por la mañana al despertarse, ya que durante la fase de sueño la renovación de la lágrima es nula. También al finalizar el día los síntomas se agudizan induciendo además una sensación de fatiga visual. La presencia de “legañas” se evidencia por las mañanas. La disminución de agudeza visual relatada por los pacientes suele ser leve y presenta la curiosidad de que mejora con el parpadeo. El síndrome de ojo seco también induce una hipersensibilidad a la luz y un deslumbramiento superior al [habitual.] |
1.3.1. Mucosa oral
Los síntomas relacionados con la xerostomía suelen iniciarse de forma insidiosa (Figura 3). Según el grado de intensidad de afección, el paciente puede presentar dificultad para hablar y comer (especialmente alimentos sólidos) y como consecuencia, perder peso, presentar halitosis, alteración del sabor de los alimentos, disestesias, sensación de ardor o quemazón bucal y labial e intolerancia para alimentos ácidos. En casos graves, el paciente suele llevar consigo una botella de agua para conseguir un alivio rápido de los síntomas. Con la hiposecreción salival se pierde el efecto tampón de la saliva y se incrementa la aparición de caries por el predominio de la flora favorecedora de la lesión dental (Streptococcus mutans), que puede agravarse con la presencia de enfermedad periodontal asociada. La hiposecreción salival conduce también al aumento de infecciones bucales, especialmente por Candida albicans (51). En los pacientes con SS, la prevalencia de candidiasis oral alcanza el 70%. Se manifiesta con síntomas de ardor y quemazón bucal, además de intolerancia a los alimentos ácidos y picantes. [página 41] [...] 1.3.2. Mucosa ocular Aparece en más del 90% de los pacientes y es la manifestación clínica más frecuente del SS (Figura 4). La expresión que refieren es la sensación constante de tener arena o tierra en lo ojos. Las molestias son más intensas por la mañana al despertarse, ya que durante la fase de sueño la renovación de la lágrima es nula. También al finalizar el día los síntomas se agudizan induciendo además una sensación de fatiga visual. La presencia de “legañas” se evidencia por las mañanas. La disminución de agudeza visual relatada por los pacientes suele ser leve y presenta la curiosidad de que mejora con el parpadeo. El síndrome de ojo seco también induce una hipersensibilidad a la luz y un deslumbramiento superior al habitual. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [9.] Mpl/Fragment 039 01 - Diskussion Bearbeitet: 27. November 2014, 17:30 PlagProf:-) Erstellt: 8. November 2014, 19:34 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 39, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 41, 42, 43, Zeilen: 41: 11ss.; 42: 1 ss.; 43: 1ss. |
|---|---|
| Relatan no poder prescindir de las gafas de sol al salir a la calle, debido a la irritación permanente de las terminaciones nerviosas del epitelio corneal. Cuando la fotofobia es tan intensa que dificulta o imposibilita la apertura palpebral o bien existe dolor, debemos sospechar la aparición de úlceras corneales. Finalmente, la disminución de la formación de la película lagrimal se hace evidente en los usuarios de lentes de contacto por empezar a presentar síntomas de intolerancia a las mismas.
1.3.3. Afección de otras mucosas La sequedad de mucosas ocurre en la práctica totalidad de los pacientes a nivel de la faringe, además de la boca. Aunque frecuente, la lesión es leve y tiene poca importancia clínica en la mayoría de los casos. La sintomatología más frecuente que pueden presentar los pacientes son el prurito y la sensación de cuerpo extraño faríngeo secundaria a la sequedad de la mucosa faringolaríngea. Estos síntomas provocan frecuentemente carraspeo y tos seca. La sequedad faríngea provoca también problemas de disfagia que aparece aproximadamente en el 30% de los pacientes. A nivel laríngeo, el SS puede progresar hacia una laringitis crónica en la cual la mucosa laríngea está persistentemente seca y consecuentemente conduce a la atrofia. La afección de otras mucosas origina una amplia variedad de manifestaciones clínicas: síntomas atribuibles a la sequedad de la mucosa respiratoria (sequedad nasal, epistaxis o tos irritativa), sequedad cutánea (xerosis) secundaria a la disminución en la producción de sudor por la infiltración de las glándulas exocrinas, con sequedad y caída del cabello, y sequedad vulvovaginal con prurito vaginal y dispareunia en las mujeres que padecen la enfermedad. |
Relatan no poder prescindir de las gafas de sol al salir a la calle, debido a la irritación permanente de las terminaciones nerviosas del epitelio corneal. Cuando la fotofobia es tan intensa que dificulta o imposibilita la apertura palpebral o bien existe dolor, debemos sospechar la aparición de úlceras corneales. Finalmente, la disminución de la formación de la
[página 42] película lagrimal se hace evidente en los usuarios de lentes de contacto por empezar a presentar síntomas de intolerancia a las mismas. [...] 1.3.3. Afección de otras mucosas La sequedad de mucosas ocurre en la práctica totalidad de los pacientes a nivel de la faringe, además de la boca. Aunque frecuente, la lesión es leve y tiene poca importancia clínica en la mayoría de los casos. La sintomatología más frecuente que pueden presentar los pacientes son el prurito y la sensación de cuerpo extraño faríngeo secundaria a la sequedad de la mucosa faringolaríngea. Estos síntomas provocan frecuentemente carraspeo y tos seca. La sequedad faríngea provoca también problemas de disfagia que aparece aproximadamente en el 30% de los pacientes. A nivel laríngeo, el SS puede progresar hacia una [página 43] laringitis crónica en la cual la mucosa laríngea está persistentemente seca y consecuentemente conduce a la atrofia. La afección de otras mucosas origina una amplia variedad de manifestaciones clínicas: síntomas atribuibles a la sequedad de la mucosa respiratoria (sequedad nasal, epistaxis o tos irritativa), sequedad cutánea (xerosis) secundaria a la disminución en la producción de sudor por la infiltración de las glándulas exocrinas, con sequedad y caída del cabello, y sequedad vulvovaginal con prurito vaginal y dispareunia en las mujeres que padecen la enfermedad. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [10.] Mpl/Fragment 040 01 - Diskussion Bearbeitet: 27. November 2014, 21:17 Hindemith Erstellt: 8. November 2014, 19:37 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 40, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 43, 44, Zeilen: 43: 10 ss.; 44: 1 ss. |
|---|---|
| 1.3.4. Afección general
La sintomatología derivada de la afección general que presenta el paciente con SS es diversa y suele estar presente en una gran parte de los pacientes, siendo la fatiga, los dolores generalizados, el decaimiento y el insomnio las manifestaciones más frecuentes. En muchas ocasiones todos estos síntomas se relacionan con la existencia de una Fibromialgia (FM) asociada, lo que obliga a realizar un enfoque terapéutico multidisciplinario del paciente (52). El Síndrome de Fatiga Crónica (SFC) es otro proceso que cursa con afección general y que no tiene un tratamiento efectivo a largo plazo (53). Algunos estudios sugieren una mayor frecuencia de procesos alérgicos en los pacientes con SS. Antonen et al (54) han detectado una mayor frecuencia de alergias a diversos antibióticos, mientras que Tishler et al (55) han detectado una mayor presencia de reacciones alérgicas farmacológicas y de contacto, especialmente en los pacientes con anticuerpos anti-Ro/SSA. Se ha descrito fiebre intermitente hasta en un 40% de pacientes con SS, aunque sin grandes alteraciones en las proteínas de fase aguda como la proteína C reactiva (PCR) (a diferencia de la fiebre de origen infeccioso). La fiebre podría estar originada por elevados niveles de citocinas circulantes que reflejarían la actividad inmunoinflamatoria del SS. Su persistencia obliga a descartar procesos linfoproliferativos. 1.3.5. Piel Las manifestaciones cutáneas del SS incluyen púrpura, eritema anular, erupciones similares al eritema multiforme, liquen plano, eritema nodoso o xerosis, entre otras. Las lesiones cutáneas del SS, especialmente la púrpura y el eritema anular, se asocian a la [presencia de anti-Ro/SSA y anti-La/SSB.] |
1.3.4. Afección general
La sintomatología derivada de la afección general que presenta el paciente con SS es diversa y suele estar presente en una gran parte de los pacientes, siendo la fatiga, los dolores generalizados, el decaimiento y el insomnio las manifestaciones más frecuentes. En muchas ocasiones todos estos síntomas se relacionan con la existencia de una Fibromialgia (FM) asociada, lo que obliga a realizar un enfoque terapéutico multidisciplinario del paciente (52). El Síndrome de Fatiga Crónica (SFC) es otro proceso que cursa con afección general y que no tiene un tratamiento efectivo a largo plazo (53). Algunos estudios sugieren una mayor frecuencia de procesos alérgicos en los pacientes con SS. Antonen et al (54) han detectado una mayor frecuencia de alergias a diversos antibióticos, mientras que Tishler et al (55) han detectado una mayor presencia de reacciones alérgicas farmacológicas y de contacto, especialmente en los pacientes con anticuerpos anti-Ro/SS-A. [página 44] Se ha descrito fiebre intermitente hasta en un 40% de pacientes con SS, aunque sin grandes alteraciones en las proteínas de fase aguda como la proteína C reactiva (PCR) (a diferencia de la fiebre de origen infeccioso). La fiebre podría estar originada por elevados niveles de citocinas circulantes que reflejarían la actividad inmunoinflamatoria del SS. Su persistencia obliga a descartar procesos linfoproliferativos. 1.3.5. Piel Las manifestaciones cutáneas del SS incluyen púrpura, eritema anular, erupciones similares al eritema multiforme, liquen plano, eritema nodoso o xerosis, entre otras. Las lesiones cutáneas del SS, especialmente la púrpura y el eritema anular, se asocian a la presencia de anti-Ro/SSA y anti-La/SSB. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [11.] Mpl/Fragment 041 01 - Diskussion Bearbeitet: 27. November 2014, 19:59 PlagProf:-) Erstellt: 8. November 2014, 19:39 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 41, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 44, 45, Zeilen: 44: 11 ss.; 45: 1 ss. |
|---|---|
| Por otra parte, las mujeres con SS pueden tener hijos con lupus eritematoso neonatal, detectándose en todos ellos la presencia de anticuerpos anti-Ro/SSA.
La manifestación más frecuente de vasculitis cutánea en el SSp es la púrpura palpable, generalmente en las extremidades inferiores. La segunda afección cutánea por frecuencia es la urticaria-vasculitis. De forma característica, estas lesiones eritematosas generalmente persisten más de 24 horas, a diferencia de la urticaria clásica cuya duración es menor de 4-6 horas. Generalmente no son pruriginosas, aunque pueden producir una sensación de “quemazón”. Otras manifestaciones cutáneas incluyen máculas eritematosas y lesiones papulares. Con menos frecuencia se observan nódulos subcutáneos, infartos digitales, úlceras y gangrena. El substrato histológico es una vasculitis leucocitoclástica, aunque se han descrito casos de vasculitis necrotizante de mediano vaso (56). Se han descrito otras lesiones cutáneas como el eritema anular, la paniculitis, el eritema nodoso, el síndrome de Sweet o el liquen plano en el SS (56). 1.3.6. Aparato locomotor Las manifestaciones articulares más frecuentes son las poliartralgias, aunque se puede observar poliartritis no erosiva o bien una oligoartritis persistente. Las articulaciones más frecuentemente afectadas son las rodillas, las metacarpofalángicas y las interfalángicas proximales. Los síntomas articulares pueden preceder, coincidir o aparecer durante el transcurso de la enfermedad y no parece existir asociación entre las manifestaciones articulares y el resto de alteraciones clínicas o inmunológicas. También con mucha frecuencia el paciente con SS presenta dolores generalizados y FM asociada. |
Por otra parte, las mujeres con SS pueden tener hijos con lupus eritematoso neonatal, detectándose en todos ellos la presencia de anticuerpos anti-Ro/SS-A.
La manifestación más frecuente de vasculitis cutánea en el SSp es la púrpura palpable, generalmente en las extremidades inferiores (Figura 5). La segunda afección cutánea por frecuencia es la urticaria-vasculitis. De forma característica, estas lesiones eritematosas generalmente persisten más de 24 horas, a diferencia de la urticaria clásica cuya duración es menor de 4-6 horas. Generalmente no son pruriginosas, aunque pueden producir una sensación de “quemazón”. Otras manifestaciones cutáneas incluyen máculas eritematosas y lesiones papulares. Con menos frecuencia se observan nódulos subcutáneos, infartos digitales, úlceras y gangrena. El substrato histológico es una vasculitis leucocitoclástica, aunque se han descrito casos de vasculitis necrotizante de mediano vaso (56). [página 45] [...] Se han descrito otras lesiones cutáneas como el eritema anular, la paniculitis, el eritema nodoso, el síndrome de Sweet o el liquen plano en el SS (56). 1.3.6. Aparato locomotor Las manifestaciones articulares más frecuentes son las poliartralgias, aunque se puede observar poliartritis no erosiva o bien una oligoartritis persistente. Las articulaciones más frecuentemente afectadas son las rodillas, las metacarpofalángicas y las interfalángicas proximales. Los síntomas articulares pueden preceder, coincidir o aparecer durante el transcurso de la enfermedad y no parece existir asociación entre las manifestaciones articulares y el resto de alteraciones clínicas o inmunológicas. También con mucha frecuencia el paciente con SS presenta dolores generalizados y FM asociada. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [12.] Mpl/Fragment 042 01 - Diskussion Bearbeitet: 27. November 2014, 20:04 PlagProf:-) Erstellt: 8. November 2014, 19:41 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 42, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 46, 47, Zeilen: 46: 1ss. ; 47: 1 ss. |
|---|---|
| La frecuencia de la afección muscular oscila entre el 0 y el 9%, siendo las mialgias el síntoma más frecuente. Como procesos asociados que induzcan afección muscular en un paciente con SS destacan la asociación con una miopatía inflamatoria, miositis de causa farmacológica o miopatía con cuerpos de inclusión (IBM). Otros procesos como la miositis focal, granulomatosa o vasculítica suelen cursar de manera silente. Recientemente, Lindvall et al (57) analizaron la afección muscular en 48 pacientes con SS, y encontraron que el 44% manifestaban mialgias. Se realizaron 36 biopsias musculares, de las que el 72% mostraron signos inflamatorios. Sólo 5 (10%) pacientes presentaron datos clínicos e histológicos sugestivos de polimiositis y 8 datos histológicos compatibles con IBM (aunque ninguno de estos pacientes presentaba datos clínicos de miopatía).
1.3.7. Aparato respiratorio La disfunción y la atrofia de las glándulas mucosas que recubren el árbol traqueobronquial originan una disminución de la secreción mucosa, aumento de su viscosidad y defectos en el aclaramiento mucociliar. La principal consecuencia es la formación de tapones de moco en los bronquios, que ocasionan sobreinfecciones posteriores, y disminución del surfactante alveolar con producción de atelectasias laminares. La denominada “bronquitis sicca” se considera la principal afección respiratoria del paciente con SS, y suele manifestarse como una enfermedad pulmonar de pequeñas vías aéreas. Los estudios sobre el funcionalismo pulmonar muestran resultados dispares. La presencia de una hipersensibilidad o hiperrreactividad bronquial se ha descrito en el 42- 60% de los pacientes con SS, mientras que la afección obstructiva varía desde la ausencia hasta el 50% (58). Se han realizado pocos estudios prospectivos, con resultados contradictorios, aunque la mayoría (59-61) no ha encontrado ningún cambio significativo [en el funcionalismo respiratorio en pacientes seguidos entre 2 y 10 años.] |
La frecuencia de la afección muscular oscila entre el 0 y el 9%, siendo las mialgias el síntoma más frecuente. Como procesos asociados que induzcan afección muscular en un paciente con SS destacan la asociación con una miopatía inflamatoria, miositis de causa farmacológica o miopatía con cuerpos de inclusión (IBM). Otros procesos como la miositis focal, granulomatosa o vasculítica suelen cursar de manera silente. Recientemente, Lindvall et al (57) analizaron la afección muscular en 48 pacientes con SS, y encontraron que el 44% manifestaban mialgias. Se realizaron 36 biopsias musculares, de las que el 72% mostraron signos inflamatorios. Sólo 5 (10%) pacientes presentaron datos clínicos e histológicos sugestivos de polimiositis y 8 datos histológicos compatibles con IBM (aunque ninguno de estos pacientes presentaba datos clínicos de miopatía).
1.3.7. Aparato respiratorio La disfunción y la atrofia de las glándulas mucosas que recubren el árbol traqueobronquial originan una disminución de la secreción mucosa, aumento de su viscosidad y defectos en el aclaramiento mucociliar. La principal consecuencia es la formación de tapones de moco en los bronquios, que ocasionan sobreinfecciones posteriores, y disminución del surfactante alveolar con producción de atelectasias laminares. La denominada “bronquitis sicca” se considera la principal afección respiratoria del paciente con SS, y suele manifestarse como una enfermedad pulmonar de pequeñas vías aéreas. Los estudios sobre el funcionalismo pulmonar muestran resultados dispares. La presencia de una hipersensibilidad o hiperrreactividad bronquial se ha descrito en el 42-60% de los pacientes con SS, mientras que la afección [página 47] obstructiva varía desde la ausencia hasta el 50% (58). Se han realizado pocos estudios prospectivos, con resultados contradictorios, aunque la mayoría (59-61) no ha encontrado ningún cambio significativo en el funcionalismo respiratorio en pacientes seguidos entre 2 y 10 años. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [13.] Mpl/Fragment 043 01 - Diskussion Bearbeitet: 27. November 2014, 20:08 PlagProf:-) Erstellt: 8. November 2014, 19:43 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 43, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 47, 48, Zeilen: 47: 4 ss.; 48: 1 ss. |
|---|---|
| En otros estudios no se han detectado diferencias significativas en la capacidad de difusión del monóxido de carbono (DLCO) entre pacientes con SSp y controles. Los resultados de estos estudios deben interpretarse con cautela, debido a que la gran mayoría se han realizado en series pequeñas de pacientes, posiblemente con una gran heterogeneidad en sus patologías pulmonares (58).
La afección pulmonar intersticial puede observarse en el 5-10% de pacientes. La forma más frecuente es la neumonitis intersticial linfocítica. Algunos pacientes presentan un curso benigno con resolución o estabilización. Hatron et al (62) realizaron lavado broncoalveolar (LBA) a pacientes con SSp y SS secundario (con o sin síntomas respiratorios), demostrando que hasta el 50% de los pacientes con SSp asintomáticos y sin alteraciones radiológicas de enfermedad pulmonar tenía un LBA anormal (de predominio linfocítico en un 69% de casos). La afección pleural no ha sido considerada como una manifestación extraglandular típica del SSp, por lo que su presencia hace sospechar la asociación con otra enfermedad autoinmune sistémica (especialmente LES) o bien neumonías recurrentes y atelectasias. Sin embargo, se han descrito casos aislados en los que la afección pleural ha constituido la primera manifestación de un SSp tras excluirse otras enfermedades autoinmunes de base (63). La hipertensión pulmonar constituye una manifestación poco frecuente del SS (64), aunque se han descrito en la literatura varios casos aislados sin que exista una enfermedad pulmonar de base. En la mayoría de estos casos el curso de la enfermedad suele ser mortal. La bronquiolitis obliterante con o sin neumonía organizada (65), es también una manifestación muy poco frecuente del SS. Se ha descrito también la asociación excepcional con amiloidosis pulmonar (66) y “shrinking lung” (67). |
En otros estudios no se han detectado diferencias significativas en la capacidad de difusión del monóxido de carbono (DLCO) entre pacientes con SSp y controles. Los resultados de estos estudios deben interpretarse con cautela, debido a que la gran mayoría se han realizado en series pequeñas de pacientes, posiblemente con una gran heterogeneidad en sus patologías pulmonares (58).
La afección pulmonar intersticial puede observarse en el 5-10% de pacientes. La forma más frecuente es la neumonitis intersticial linfocítica (Figura 6). Algunos pacientes presentan un curso benigno con resolución o estabilización. Hatron et al (62) realizaron lavado broncoalveolar (LBA) a pacientes con SSp y SS secundario (con o sin síntomas respiratorios), demostrando que hasta el 50% de los pacientes con SSp asintomáticos y sin alteraciones radiológicas de enfermedad pulmonar tenía un LBA anormal (de predominio linfocítico en un 69% de casos). La afección pleural no ha sido considerada como una manifestación extraglandular típica del SSp, por lo que su presencia hace sospechar la asociación con otra enfermedad autoinmune sistémica (especialmente LES) o bien neumonías recurrentes y atelectasias. Sin embargo, se han descrito casos aislados en los que la afección pleural ha constituido la primera manifestación de un SSp tras excluirse otras enfermedades autoinmunes de base (63). La hipertensión pulmonar constituye una manifestación poco frecuente del SS (64), aunque se han descrito en la literatura varios casos aislados sin que exista una [página 48] enfermedad pulmonar de base. En la mayoría de estos casos el curso de la enfermedad suele ser mortal. La bronquiolitis obliterante con o sin neumonía organizada (65), es también una manifestación muy poco frecuente del SS. Se ha descrito también la asociación excepcional con amiloidosis pulmonar (66) y “shrinking lung” (67). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [14.] Mpl/Fragment 044 01 - Diskussion Bearbeitet: 27. November 2014, 20:11 PlagProf:-) Erstellt: 8. November 2014, 19:46 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 44, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 48, 49, Zeilen: 48: 6ss.; 49: 1 ss. |
|---|---|
| 1.3.8. Afección cardiovascular
La afección cardiaca en el SS es poco frecuente. Mediante ecocardiografía puede detectarse derrame pericárdico moderado hasta en un 30% de casos, generalmente asintomático y sin repercusión hemodinámica. Gyöngyösi et al (68) han descrito un solo caso de pericarditis aguda exudativa en un total de 64 pacientes, mientras que se observaron engrosamientos ecogénicos del pericardio en 21 enfermos (33%), todos ellos asintomáticos. Rantapää-Dahlqvist et al (69) describieron, en una serie de 27 enfermos con SS y examen clínico cardiológico, la presencia de datos ecocardiográficos de pericarditis en 9 de ellos (33%) y de éstos, 4 habían presentado síntomas torácicos. Dada la escasa frecuencia de derrame pericárdico en el SSp, su presencia obliga a descartar la presencia de otras enfermedades autoinmunes o neoplásicas asociadas. Asimismo, como consecuencia de la fibrosis pulmonar puede producirse "cor pulmonale" crónico. Es excepcional la presencia de miocarditis (70). El fenómeno de Raynaud se observa en el 20% de los casos, y su evolución suele ser benigna, produciendo raramente secuelas vasculares importantes (71). En 1998, Andonopoulos et al (72) encontraron que el 70% de pacientes con SS presentaban alteración en diversas pruebas cardiovasculares, aunque estudios posteriores no han confirmado esta elevada proporción (73,74). 1.3.9. Afección digestiva El tubo digestivo está cubierto por una mucosa con múltiples glándulas exocrinas cuya lesión puede originar diversas manifestaciones clínicas. La incidencia real de afección esofágica en el SS no se conoce, aunque probablemente sea mayor de lo que la clínica indica. La disfagia, definida como una dificultad en la deglución, es frecuente en pacientes con SSp, y suele estar relacionada con la existencia de una xerostomía severa, [aunque en ocasiones también puede ser indicativa de una afección intrínseca del esófago.] |
1.3.8. Afección cardiovascular
La afección cardiaca en el SS es poco frecuente. Mediante ecocardiografía puede detectarse derrame pericárdico moderado hasta en un 30% de casos, generalmente asintomático y sin repercusión hemodinámica. Gyöngyösi et al (68) han descrito un solo caso de pericarditis aguda exudativa en un total de 64 pacientes, mientras que se observaron engrosamientos ecogénicos del pericardio en 21 enfermos (33%), todos ellos asintomáticos. Rantapää-Dahlqvist et al (69) [página 49] describieron, en una serie de 27 enfermos con SS y examen clínico cardiológico, la presencia de datos ecocardiográficos de pericarditis en 9 de ellos (33%) y de éstos, 4 habían presentado síntomas torácicos. Dada la escasa frecuencia de derrame pericárdico en el SSp, su presencia obliga a descartar la presencia de otras enfermedades autoinmunes o neoplásicas asociadas. Asimismo, como consecuencia de la fibrosis pulmonar puede producirse "cor pulmonale" crónico. Es excepcional la presencia de miocarditis (70). El fenómeno de Raynaud se observa en el 20% de los casos, y su evolución suele ser benigna, produciendo raramente secuelas vasculares importantes (71). En 1998, Andonopoulos et al (72) encontraron que el 70% de pacientes con SS presentaban alteración en diversas pruebas cardiovasculares, aunque estudios posteriores no han confirmado esta elevada proporción (73,74). 1.3.9. Afección digestiva El tubo digestivo está cubierto por una mucosa con múltiples glándulas exocrinas cuya lesión puede originar diversas manifestaciones clínicas. La incidencia real de afección esofágica en el SS no se conoce, aunque probablemente sea mayor de lo que la clínica indica. La disfagia, definida como una dificultad en la deglución, es frecuente en pacientes con SSp, y suele estar relacionada con la existencia de una xerostomía severa, aunque en ocasiones también puede se [sic] indicativa de una afección intrínseca del esófago. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [15.] Mpl/Fragment 045 01 - Diskussion Bearbeitet: 27. November 2014, 20:19 PlagProf:-) Erstellt: 8. November 2014, 20:21 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 45, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 49, 50, Zeilen: 49: 17 ss.; 50: 1 ss. |
|---|---|
| [La disfagia, definida como una dificultad en la deglución, es frecuente en pacientes con SSp, y suele estar relacionada con la existencia de una xerostomía severa,] aunque en ocasiones también puede ser indicativa de una afección intrínseca del esófago. Cuando existe una disfagia por afección esofágica en un paciente con SS, suele estar relacionada con alteraciones de la motilidad del esófago en relación a la coexistencia de una esclerodermia, aunque las formas primarias del SS también pueden presentar alteraciones de la motilidad esofágica. Tsianos et al (75) detectaron alteraciones de la motilidad esofágica en el 75% de los pacientes con SSp. Otro estudio realizado en pacientes con SSp (76), demostró que un 11% de ellos presentaban una afección del tramo proximal y un 22% del tramo distal. También se han descrito casos de acalasia y membranas esofágicas en pacientes con SSp (77).
Los estudios con fibrogastroscopia han revelado con relativa frecuencia la presencia de gastritis. A pesar de la existencia de una gastritis crónica, la anemia perniciosa ocurre sólo en el 3% de los casos. Las lesiones más comúnmente descritas son la gastritis crónica atrófica (78) con o sin anemia perniciosa asociada y el linfoma gástrico (79). La afección intestinal en el SS no ha sido bien definida y se han descrito algunos casos de malabsorción y asociación con enfermedad celíaca. El intestino delgado y el colon pueden presentar infiltración inflamatoria. 1.3.10. Páncreas No se conoce con exactitud la prevalencia de enfermedad pancreática en pacientes con SS. Aunque cerca del 50% de pacientes con SS puede presentar insuficiencia pancreática exocrina, la mayoría están asintomáticos. Los signos y síntomas, en caso de existir, siempre estarán circunscritos al páncreas exocrino. No suele existir dolor abdominal, exceptuando en los casos raros de vasculitis pancreática asociada, ni esteatorrea. En ocasiones, las determinaciones séricas de las enzimas pancreáticas son útiles cuando se sospecha una afección pancreática incipiente (80-82), que puede [manifestarse como una pancreatitis aguda o una pancreatitis crónica subclínica (83,84).] |
La disfagia, definida como una dificultad en la deglución, es frecuente en pacientes con SSp, y suele estar relacionada con la existencia de una xerostomía severa, aunque en ocasiones también puede se [sic] indicativa de una afección intrínseca del esófago. Cuando existe una disfagia por afección esofágica en un paciente con SS, suele estar relacionada con alteraciones de la motilidad del esófago en relación a la coexistencia de una esclerodermia, aunque las formas primarias del SS también pueden presentar alteraciones de la motilidad esofágica. Tsianos et al. (75)
[página 50] detectaron alteraciones de la motilidad esofágica en el 75% de los pacientes con SSp. Otro estudio realizado en pacientes con SSp (76), demostró que un 11% de ellos presentaban una afección del tramo proximal y un 22% del tramo distal. También se han descrito casos de acalasia y membranas esofágicas en pacientes con SSp (77). Los estudios con fibrogastroscopia han revelado con relativa frecuencia la presencia de gastritis. A pesar de la existencia de una gastritis crónica, la anemia perniciosa ocurre sólo en el 3% de los casos. Las lesiones más comúnmente descritas son la gastritis crónica atrófica (78) con o sin anemia perniciosa asociada y el linfoma gástrico (79). La afección intestinal en el SS no ha sido bien definida y se han descrito algunos casos de malabsorción y asociación con enfermedad celíaca. El intestino delgado y el colon pueden presentar infiltración inflamatoria. 1.3.10. Páncreas No se conoce con exactitud la prevalencia de enfermedad pancreática en pacientes con SS. Aunque cerca del 50% de pacientes con SS puede presentar insuficiencia pancreática exocrina, la mayoría están asintomáticos. Los signos y síntomas, en caso de existir, siempre estarán circunscritos al páncreas exocrino. No suele existir dolor abdominal, exceptuando en los casos raros de vasculitis pancreática asociada, ni esteatorrea. En ocasiones, las determinaciones séricas de las enzimas pancreáticas son útiles cuando se sospecha una afección pancreática incipiente (80-82), que puede manifestarse como una pancreatitis aguda o una pancreatitis crónica subclínica (83,84). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [16.] Mpl/Fragment 046 01 - Diskussion Bearbeitet: 27. November 2014, 20:22 PlagProf:-) Erstellt: 8. November 2014, 20:27 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 46, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 50, 51, 52, Zeilen: 50: 20 ss.; 51: 1 ss. ; 52: 1 ss. |
|---|---|
| [En ocasiones, las determinaciones séricas de las enzimas pancreáticas son útiles cuando se sospecha una afección pancreática incipiente (80-82), que puede] manifestarse como una pancreatitis aguda o una pancreatitis crónica subclínica (83,84). Puede presentarse tanto en las formas primarias como secundarias del SS aunque las manifestaciones clínicas típicas de la afección pancreática son poco habituales y no suelen acompañarse de insuficiencia pancreática exocrina. Los porcentajes varían según las técnicas utilizadas para el estudio de la función exocrina pancreática, siendo la alteración más comúnmente descrita, la disminución del volumen de secreción pancreática, utilizando la prueba de estimulación con secretina, o el test secretina-pancreozimina (85).
1.3.11. Hígado y vías biliares Cuando se detecta alteración clínica o biológica hepática en pacientes con SS debe analizarse si existe alguna otra enfermedad asociada, en primer lugar la infección por el VHC, que podría afectar, según el área geográfica, al 5-10% de pacientes diagnosticados de SS “primario”. La enfermedad hepática más comúnmente asociada al SS es la cirrosis biliar primaria (CBP), que se caracteriza por un infiltrado linfocitario en los espacios porta, alrededor de los conductos biliares lesionados, y una obstrucción fibrosa de los conductos biliares intrahepáticos. En más del 90% de los pacientes se detectan anticuerpos antimitocondriales (AMA). El síntoma inicial suele ser el prurito, desarrollándose ictericia a lo largo de meses o años. La CBP se asocia al SS con una incidencia muy alta, incluso superior a la conocida asociación del SS con la AR. Diversos estudios indican que entre un 40 y un 75% de pacientes con CBP presentan un SS asociado (86-88). La clínica y el perfil serológico de los pacientes con SS y afección hepática, una vez excluida la CBP y la infección por VHC, suele ser parecida a la hepatitis autoinmune (HAI) tipo 1 (89,90). No se conoce la frecuencia de asociación de la HAI con el SS, pero probablemente sea muy inferior a la descrita en la CBP, ya que las descripciones sobre [HAI y SS son excepcionales, aunque su identificación es importante dado el efecto beneficioso del tratamiento con glucocorticoides (91).] |
En ocasiones, las determinaciones séricas de las enzimas pancreáticas son útiles cuando se sospecha una afección pancreática incipiente (80-82), que puede manifestarse como una pancreatitis aguda o una pancreatitis crónica subclínica (83,84). Puede presentarse tanto en las formas primarias como secundarias del SS aunque las manifestaciones
[página 51] clínicas típicas de la afección pancreática son poco habituales y no suelen acompañarse de insuficiencia pancreática exocrina. Los porcentajes varían según las técnicas utilizadas para el estudio de la función exocrina pancreática, siendo la alteración más comúnmente descrita, la disminución del volumen de secreción pancreática, utilizando la prueba de estimulación con secretina, o el test secretina-pancreozimina (85). 1.3.11. Hígado y vías biliares Cuando se detecta alteración clínica o biológica hepática en pacientes con SS debe analizarse si existe alguna otra enfermedad asociada, en primer lugar la infección por el VHC, que podría afectar, según el área geográfica, al 5-10% de pacientes diagnosticados de SS “primario”. La enfermedad hepática más comúnmente asociada al SS es la cirrosis biliar primaria (CBP), que se caracteriza por un infiltrado linfocitario en los espacios porta, alrededor de los conductos biliares lesionados, y una obstrucción fibrosa de los conductos biliares intrahepáticos. En más del 90% de los pacientes se detectan anticuerpos antimitocondriales (AMA). El síntoma inicial suele ser el prurito, desarrollándose ictericia a lo largo de meses o años. La CBP se asocia al SS con una incidencia muy alta, incluso superior a la conocida asociación del SS con la AR. Diversos estudios indican que entre un 40 y un 75% de pacientes con CBP presentan un SS asociado (86-88). La clínica y el perfil serológico de los pacientes con SS y afección hepática, una vez excluida la CBP y la infección por VHC, suele ser parecida a la hepatitis autoinmune (HA) tipo 1 (89,90). No se conoce la frecuencia de asociación de la HA con el SS, pero probablemente sea muy inferior a la descrita [página 52] en la CBP, ya que las descripciones sobre HA y SS son excepcionales, aunque su identificación es importante dado el efecto beneficioso del tratamiento con glucocorticoides (91). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [17.] Mpl/Fragment 047 01 - Diskussion Bearbeitet: 27. November 2014, 20:27 PlagProf:-) Erstellt: 8. November 2014, 20:30 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 47, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 51, 52, 53, Zeilen: 51: 23 ss.; 52: 1 ss.; 53: 1 ss. |
|---|---|
| [No se conoce la frecuencia de asociación de la HAI con el SS, pero probablemente sea muy inferior a la descrita en la CBP, ya que las descripciones sobre] HAI y SS son excepcionales, aunque su identificación es importante dado el efecto beneficioso del tratamiento con glucocorticoides (91).
La colangitis autoinmune (CA) se caracteriza por presentar datos clínicos y de laboratorio de colestasis en presencia de AMA negativos y anticuerpos antinucleares (ANA) positivos. Las características histológicas e inmunohistoquímicas son muy parecidas a las de la CBP y se ha sugerido que la CA sería un subgrupo de CBP con AMA negativos. La colangitis esclerosante primaria (CEP) es un proceso inflamatorio, esclerosante y obliterante que puede afectar a los conductos biliares extra o intrahepáticos. Se asocia en un 70% con la enfermedad inflamatoria intestinal (EII), fundamentalmente colitis ulcerosa (CU), aunque se ha descrito la asociación de CEP, pancreatitis crónica y SS (92). Además, los pacientes con SSp pueden presentar de forma excepcional, otros tipos de hepatopatías, como atrofia lobar hepática e hiperplasia nodular regenerativa (93). 1.3.12. Riñones La incidencia de afección renal varía según los distintos autores, desde el 18.4% al 67% (94,95). De todas formas, recientes series que incluyen más de 400 pacientes muestran una prevalencia del 4-6% (96,97). La alteración más frecuentemente descrita es la nefritis intersticial (98) seguido de la glomerulonefritis (GMN) (99). La afección tubulointersticial suele manifestarse como una acidosis tubular renal distal (ATRd), y se presenta la mayoría de las veces sin manifestaciones clínicas evidentes (100), aunque puede incrementar el riesgo de desarrollar litiasis renal y de forma excepcional nefrocalcinosis (101). En series más recientes, se ha descrito un 11-33% de ATRd, siendo la forma incompleta el tipo de presentación más frecuente (102,103). En la mayoría de pacientes, la acidosis solo se manifiesta después de la sobrecarga con cloruro amónico (104). |
No se conoce la frecuencia de asociación de la HA con el SS, pero probablemente sea muy inferior a la descrita
[página 52] en la CBP, ya que las descripciones sobre HA y SS son excepcionales, aunque su identificación es importante dado el efecto beneficioso del tratamiento con glucocorticoides (91). La colangitis autoinmune (CA) se caracteriza por presentar datos clínicos y de laboratorio de colestasis en presencia de AMA negativos y anticuerpos antinucleares (ANA) positivos. Las características histológicas e inmunohistoquímicas son muy parecidas a las de la CBP y se ha sugerido que la CA sería un subgrupo de CBP con AMA negativos. La colangitis esclerosante primaria (CEP) es un proceso inflamatorio, esclerosante y obliterante que puede afectar a los conductos biliares extra o intrahepáticos. Se asocia en un 70% con la enfermedad inflamatoria intestinal (EII), fundamentalmente colitis ulcerosa (CU), aunque se ha descrito la asociación de CEP, pancreatitis crónica y SS (92). Además, los pacientes con SSp pueden presentar de forma excepcional, otros tipos de hepatopatías, como atrofia lobar hepática e hiperplasia nodular regenerativa (93). 1.3.12. Riñones La incidencia de afección renal varía según los distintos autores, desde el 18.4% al 67% (94,95). De todas formas, recientes series que incluyen más de 400 pacientes muestran una prevalencia del 4-6% (96,97). La alteración más frecuentemente descrita es la nefritis intersticial (98) seguido de la glomerulonefritis (GMN) (99). La afección tubulointersticial suele manifestarse como una acidosis tubular renal distal (ATRd), y se presenta la mayoría de las veces sin manifestaciones clínicas evidentes (100), aunque puede incrementar el riesgo de desarrollar litiasis [página 53] renal y de forma excepcional nefrocalcinosis (101). En series más recientes, se ha descrito un 11-33% de ATRd, siendo la forma incompleta el tipo de presentación más frecuente (102,103). En la mayoría de pacientes, la acidosis solo se manifiesta después de la sobrecarga con cloruro amónico (104). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [18.] Mpl/Fragment 048 01 - Diskussion Bearbeitet: 27. November 2014, 20:32 PlagProf:-) Erstellt: 8. November 2014, 20:33 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 48, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 53, 54, Zeilen: 53: 4 ss.; 54: 1 ss. |
|---|---|
| [La acidosis renal tubular proximal (ATRp), con o sin las manifestaciones] del síndrome de Fanconi (glucosuria, aminoaciduria, fosfaturia y uricosuria) es infrecuente en pacientes con SSp.
La GMN en pacientes con SSp obliga a descartar, en primer lugar, otras causas de GMN. Las lesiones más habitualmente observadas son la GMN mesangial, la GMN membranoproliferativa y la GMN membranosa (105,106). En el trabajo de Goules et al (96) se analizan biopsias renales de 20 pacientes con SS, observándose GMN mesangial en 5 casos, caracterizada clínicamente por discreta hematuria, proteinuria, hipertensión e insuficiencia renal moderada, y GMN membranoproliferativa en otros 4, con una combinación variable de síndrome nefrítico y nefrótico. Ocho de los pacientes presentaron CM. 1.3.13. Sistema nervioso central La prevalencia de la afección del sistema nervioso central (SNC) varía según los diversos estudios, aunque en las grandes series suele ser excepcional (inferior al 5% de casos). Los síntomas pueden ser discretos e insidiosos y la afección, tanto cerebral como medular. La afección del SNC puede causar lesiones focales (déficits motores o sensitivos de tipo hemi o monoparesias) o difusas (alteraciones de las funciones cognoscitivas, meningitis asépticas, encefalopatía o demencia progresiva asociada), y varía desde la presencia de déficit motor, afasia, disartria, amaurosis y síndrome cerebeloso, en la expresión de daño difuso como encefalopatía subaguda, meningitis aséptica, disfunción cognitiva y anormalidades neuropsiquiátricas. El sustrato histológico consiste en una vasculitis inflamatoria de pequeño y mediano vaso y, con frecuencia, se observan microinfartos y hemorragias que pueden comprometer los pequeños vasos del parénquima cerebral y meninges. La afección del SNC guarda estrecha relación con la existencia de lesiones vasculíticas en otros órganos como la piel o el músculo (107). |
La acidosis renal tubular proximal (ATRp), con o sin las manifestaciones del síndrome de Fanconi (glucosuria, aminoaciduria, fosfaturia y uricosuria) es infrecuente en pacientes con SSp.
La GMN en pacientes con SSp obliga a descartar, en primer lugar, otras causas de GMN. Las lesiones más habitualmente observadas son la GMN mesangial, la GMN membranoproliferativa y la GMN membranosa (105,106). En el trabajo de Goules et al (96) se analizan biopsias renales de 20 pacientes con SS, observándose GMN mesangial en 5 casos, caracterizada clínicamente por discreta hematuria, proteinuria, hipertensión e insuficiencia renal moderada, y GMN membranoproliferativa en otros 4, con una combinación variable de síndrome nefrítico y nefrótico. Ocho de los pacientes presentaron CM. 1.3.13. Sistema nervioso central La prevalencia de la afección del sistema nervioso central (SNC) varía según los diversos estudios, aunque en las grandes series suele ser excepcional (inferior al 5% de casos). Los síntomas pueden ser discretos e insidiosos y la afección, tanto cerebral como medular. La afección del SNC puede causar lesiones focales (déficits motores o sensitivos de tipo hemi o monoparesias) o difusas (alteraciones de las funciones cognoscitivas, meningitis asépticas, encefalopatía o demencia progresiva asociada), y varía desde la presencia de déficit motor, afasia, disartria, amaurosis y síndrome cerebeloso, en la expresión [página 54] de daño difuso como encefalopatía subaguda, meningitis aséptica, disfunción cognitiva y anormalidades neuropsiquiátricas. El sustrato histológico consiste en una vasculitis inflamatoria de pequeño y mediano vaso y, con frecuencia, se observan microinfartos y hemorragias que pueden comprometer los pequeños vasos del parénquima cerebral y meninges. La afección del SNC guarda estrecha relación con la existencia de lesiones vasculíticas en otros órganos como la piel o el músculo (107). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [19.] Mpl/Fragment 049 01 - Diskussion Bearbeitet: 27. November 2014, 20:36 PlagProf:-) Erstellt: 8. November 2014, 20:36 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 49, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 54, 55, Zeilen: 54: 6 ss.; 55: 1 ss. |
|---|---|
| [La afección] medular produce cuadros de mielitis transversa y mielopatía crónica progresiva. En el SS asociado al LES la afección del SNC es más frecuente y suele ser aguda o subaguda.
Existe además un grupo de pacientes que presentan un cuadro neurológico indistinguible de la esclerosis múltiple (EM), y recientes estudios han mostrado una mayor prevalencia de síndrome seco en pacientes con EM. Por otra parte, la presencia de lesiones de la sustancia blanca en las pruebas de imagen [tomografía axial computerizada (TAC) o resonancia magnética (RM)] es frecuente en pacientes con SSp, aunque su significado clínico no está esclarecido (108,109). La afección del SNC en el SS también incluye la existencia de migraña (109), trastornos psiquiátricos (depresión, hipocondriasis, somatización) y trastornos cognitivos con dificultad en la capacidad de concentración y atención (107,110). Las manifestaciones neuropsiquiátricas van desde trastornos afectivos como depresión o hipomanía a trastornos de la personalidad como la histeria o la psicosis, las crisis de pánico y la ansiedad entre otros casos, siendo más frecuente la depresión que ocurre hasta en el 50% de los pacientes (110). 1.3.14. Sistema nervioso periférico La afección del sistema nervioso periférico (SNP) se observa en el 10-45% de los pacientes con SS y las formas de presentación más frecuentes son la polineuropatía sensitivomotora y la neuralgia del trigémino. La presentación suele ser insidiosa, se diagnostica según los hallazgos en el electromiograma y su curso es habitualmente tórpido y con escasa respuesta al tratamiento. En las diversas series de pacientes con SSp que analizan los casos de afección del SNP, así como la forma de presentación (111), la afección del SNP se describe en el 18% de un total de 1025 pacientes estudiados, aunque las cifras oscilan ampliamente según los estudios desde el 3% al 44%. |
La afección medular produce cuadros de mielitis transversa y mielopatía crónica progresiva. En el SS asociado al LES la afección del SNC es más frecuente y suele ser aguda o subaguda.
Existe además un grupo de pacientes que presentan un cuadro neurológico indistinguible de la EM, y recientes estudios han mostrado una mayor prevalencia de síndrome seco en pacientes con EM. Por otra parte, la presencia de lesiones de la sustancia blanca en las pruebas de imagen [tomografía axial computerizada (TAC) o resonancia magnética (RM)] es frecuente en pacientes con SSp, aunque su significado clínico no está esclarecido (108,109). La afección del SNC en el SS también incluye la existencia de migraña (109), trastornos psiquiátricos (depresión, hipocondriasis, somatización) y trastornos cognitivos con dificultad en la capacidad de concentración y atención (107,110). Las manifestaciones neuropsiquiátricas van desde trastornos afectivos como depresión o hipomanía a trastornos de la personalidad como la histeria o la psicosis, las crisis de pánico y la ansiedad entre otros casos, siendo más frecuente la depresión que ocurre hasta en el 50% de los pacientes (110). [página 55] 1.3.14. Sistema nervioso periférico La afección del sistema nervioso periférico (SNP) se observa en el 10-45% de los pacientes con SS y las formas de presentación más frecuentes son la polineuropatía sensitivomotora y la neuralgia del trigémino. La presentación suele ser insidiosa, se diagnostica según los hallazgos en el electromiograma y su curso es habitualmente tórpido y con escasa respuesta al tratamiento. En las diversas series de pacientes con SSp que analizan los casos de afección del SNP, así como la forma de presentación (111), la afección del SNP se describe en el 18% de un total de 1025 pacientes estudiados, aunque las cifras oscilan ampliamente según los estudios desde el 3% al 44%. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [20.] Mpl/Fragment 050 01 - Diskussion Bearbeitet: 27. November 2014, 20:41 PlagProf:-) Erstellt: 8. November 2014, 20:38 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 50, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 55, 56, Zeilen: 55: 10 ss. ; 56: 1 ss. |
|---|---|
| En estas series predomina la afección en forma de polineuropatía mixta (41 pacientes), seguido de la neuropatía sensitiva pura (n=27), la mononeuritis múltiple (n=15) y la afección trigeminal (n=13). Son mucho menos frecuentes la neuropatía motora pura, la afección de otros pares craneales y la polirradiculopatía (111).
La polineuropatía mixta sensitivo-motora es la forma más frecuente en el SSp. Su distribución característica es distal y simétrica, generalmente con predominio de la afección sensitiva, en forma de parestesias dolorosas y disestesias,o leves hipoestesias en guante o calcetín. Suele predominar en las extremidades inferiores, es generalmente leve, no discapacitante y de tipo axonal, producida por isquemia secundaria a vasculitis en los "vasa nervorum". Por otra parte, existen dos tipos distintos de afección neurológica sensitiva, cada uno de ellos con una base etiopatogénica distinta. Puede existir una polineuropatía sensitiva pura, que cursa con hiperestesia cutánea, disestesias dolorosas y parestesias, y que es consecuencia de la afección de las pequeñas fibras nerviosas aferentes secundaria a vasculitis en los “vasa nervorum”. En segundo lugar se ha descrito la denominada neuronopatía sensitiva debida a la infiltración ganglionar linfocitaria de la raíz dorsal, también llamada neuropatía sensitiva atáxica. Esta última no es infrecuente que se manifieste antes del diagnóstico del SS, no se asocia a vasculitis sistémica y no suele responder al tratamiento con corticoides. La mayoría de las veces, es de comienzo insidioso y de lenta evolución. Consiste en la pérdida de la función sensitiva, fundamentalmente profunda (propioceptiva y vibratoria), suele iniciarse en las extremidades superiores de forma asimétrica y con progresiva afección de las extremidades inferiores (112). Se han publicado 19 casos de mononeuritis múltiple en pacientes con SS, causada por isquemia secundaria a vasculitis de los “vasa nervorum”. |
En estas series predomina la afección en forma de polineuropatía mixta (41 pacientes), seguido de la neuropatía sensitiva pura (n=27), la mononeuritis múltiple (n=15) y la afección trigeminal (n=13). Son mucho menos frecuentes la neuropatía motora pura, la afección de otros pares craneales y la polirradiculopatía (111).
La polineuropatía mixta sensitivo-motora es la forma más frecuente en el SSp. Su distribución característica es distal y simétrica, generalmente con predominio de la afección sensitiva, en forma de parestesias dolorosas y disestesias,o leves hipoestesias en guante o calcetín. Suele predominar en las extremidades inferiores, es generalmente leve, no discapacitante y de tipo axonal, producida por isquemia secundaria a vasculitis en los "vasa nervorum". Por otra parte, existen dos tipos distintos de afección neurológica sensitiva, cada uno de ellos con una base etiopatogénica distinta. Puede existir una polineuropatía sensitiva pura, que cursa con hiperestesia cutánea, disestesias dolorosas y parestesias, y que es consecuencia de la afección de las pequeñas fibras nerviosas aferentes secundaria a vasculitis en los “vasa nervorum”. En segundo [página 56] lugar se ha descrito la denominada neuronopatía sensitiva debida a la infiltración ganglionar linfocitaria de la raíz dorsal, también llamada neuropatía sensitiva atáxica. Esta última no es infrecuente que se manifieste antes del diagnóstico del SS, no se asocia a vasculitis sistémica y no suele responder al tratamiento con corticoides. La mayoría de las veces, es de comienzo insidioso y de lenta evolución. Consiste en la pérdida de la función sensitiva, fundamentalmente profunda (propioceptiva y vibratoria), suele iniciarse en las extremidades superiores de forma asimétrica y con progresiva afección de las extremidades inferiores (112). Se han publicado 19 casos de mononeuritis múltiple en pacientes con SS, causada por isquemia secundaria a vasculitis de los “vasa nervorum”. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [21.] Mpl/Fragment 051 01 - Diskussion Bearbeitet: 28. November 2014, 14:37 PlagProf:-) Erstellt: 8. November 2014, 20:40 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 51, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 56, 57, Zeilen: 56: 11 ss.; 57: 1 ss. |
|---|---|
| Finalmente, los síndromes de atrapamiento (carpal, ulnar o tarsal generalmente) son otra forma común de manifestación neurológica en el SS.
Descrita por primera vez en el SS por Kaltreider y Talal en 1969 (113), la neuropatía del trigémino es una de las afectaciones neurológicas características del SS. Se produce cuando se afecta el ganglio de Gasser. Se puede asociar a neuropatía sensitiva pura, o como única afección neurológica. Los síntomas consisten en hiperestesias o parestesias unilaterales o bilaterales en la rama maxilar y/o mandibular del nervio trigémino con función motora normal. La rama oftálmica se afecta menos frecuentemente. El dolor generalmente está presente pero no suele ser grave y es frecuente la afección del reflejo corneal. La afección del gusto, cuando ocurre suele ser ipsilateral (114). También se ha descrito la afección de otros pares craneales, de forma aislada en el caso de la afección del III par, del VII o del VIII, o bien la afección múltiple de varios pares. Las plexopatías braquiales o las polirradículoneuropatías son otros tipos de neuropatías descritos en pacientes con SS (111). 1.3.15. Otras manifestaciones a) Tiroides Diversos estudios realizados en los años 80 y 90 detectaron una prevalencia de afección tiroidea en pacientes con SS entre el 14% y el 45% (115-119). En un estudio reciente de casos y controles (120), se analizaron 160 pacientes con SSp y se detectó enfermedad tiroidea en el 36% de pacientes. Se hizo el diagnóstico de enfermedad tiroidea autoinmune en el 20% y de enfermedad tiroidea no autoinmune, principalmente hipotiroidismo, en el 16%. Sin embargo, esta prevalencia no fue significativamente mayor que en el grupo control de igual edad y sexo. |
Finalmente, los síndromes de atrapamiento (carpal, ulnar o tarsal generalmente) son otra forma común de manifestación neurológica en el SS.
Descrita por primera vez en el SS por Kaltreider y Talal en 1969 (113), la neuropatía del trigémino es una de las afectaciones neurológicas características del SS. Se produce cuando se afecta el ganglio de Gasser. Se puede asociar a neuropatía sensitiva pura, o como única afección neurológica. Los síntomas consisten en hiperestesias o parestesias unilaterales o bilaterales en la rama maxilar y/o mandibular del nervio trigémino con función motora normal. La rama oftálmica se afecta menos frecuentemente. El dolor generalmente está presente pero no suele ser grave y es frecuente la afección del reflejo corneal. La afección del gusto, cuando ocurre suele ser ipsilateral (114). También se ha descrito la afección de otros pares craneales, de forma aislada en el caso de la afección del III par, del VII o del VIII, o bien la afección múltiple de varios pares. Las plexopatías braquiales o las polirradículoneuropatías son otros tipos de neuropatías descritos en pacientes con SS (111). [página 57] 1.3.15. Otras manifestaciones a) Tiroides Diversos estudios realizados en los años 80 y 90 detectaron una prevalencia de afección tiroidea en pacientes con SS entre el 14% y el 45% (115- 119). En un estudio reciente de casos y controles (120), se analizaron 160 pacientes con SSp y se detectó enfermedad tiroidea en el 36% de pacientes. Se hizo el diagnóstico de enfermedad tiroidea autoinmune en el 20% y de enfermedad tiroidea no autoinmune, principalmente hipotiroidismo, en el 16%. Sin embargo, esta prevalencia no fue significativamente mayor que en el grupo control de igual edad y sexo. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [22.] Mpl/Fragment 052 01 - Diskussion Bearbeitet: 28. November 2014, 14:40 PlagProf:-) Erstellt: 8. November 2014, 20:42 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 52, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 57, 58, Zeilen: 57: 11 ss.; 58: 1 ss. |
|---|---|
| La enfermedad tiroidea autoinmune se observa en alrededor del 20% de pacientes con SSp, con una prevalencia de anticuerpos anti-tiroglobulina (anti-Tg) anti-peroxidasa tiroidea (anti-TPO) de hasta el 25% de pacientes. Por otra parte, otro estudio (121) encontró que hasta un tercio de los pacientes con enfermedad tiroidea autoinmune presentaban características clínicas típicas del SSp (xerostomía y xeroftalmía), lo que apoya la hipótesis de que el SSp y la enfermedad tiroidea autoinmune son 2 enfermedades estrechamente relacionadas entre sí.
El perfil clínico en más de la mitad de los pacientes con SSp y afección tiroidea es generalmente un hipotiroidismo subclínico con elevación temprana de la hormona estimuladora del tiroides (TSH). El hipertiroidismo autoinmune ha sido descrito de forma excepcional en pacientes con SSp (118,120). b) Afección otorrinolaringológica La afección del oído en las enfermedades autoinmunes no es infrecuente y en el SS está descrita tanto en la afección primaria como la que se encuentra asociada a otras enfermedades autoinmunes. Cada una de las estructuras anatómicas del oído (interno, medio y externo) puede afectarse. Ello puede traducirse en síntomas como otalgia, acúfenos, vértigo e hipoacusia, y en signos como sequedad de la piel del conducto auditivo, cera seca y ocupación del oído medio por mal funcionamiento de la trompa de Eustaquio (122). La hipoacusia que se puede observar en estos pacientes puede ser neurosensorial, de transmisión o una combinación de ambas. La hipoacusia y los acúfenos pueden aparecer aproximadamente en el 25% de los enfermos, presentándose de forma súbita en algunos de ellos. Hasta la fecha, cinco trabajos han analizado la existencia de hipoacusia neurosensorial en pacientes con SS. |
La enfermedad tiroidea autoinmune se observa en alrededor del 20% de pacientes con SSp, con una prevalencia de anticuerpos anti-tiroglobulina (anti-Tg) anti-peroxidasa tiroidea (anti-TPO) de hasta el 25% de pacientes. Por otra parte, otro estudio (121) encontró que hasta un tercio de los pacientes con enfermedad tiroidea autoinmune presentaban características clínicas típicas del SSp (xerostomía y xeroftalmía), lo que apoya la hipótesis de que el SSp y la enfermedad tiroidea autoinmune son 2 enfermedades estrechamente relacionadas entre sí.
El perfil clínico en más de la mitad de los pacientes con SSp y afección tiroidea es generalmente un hipotiroidismo subclínico con elevación temprana de la hormona estimuladora del tiroides (TSH). El hipertiroidismo autoinmune ha sido descrito de forma excepcional en pacientes con SSp (118,120). [página 58] b) Afección otorrinolaringológica La afección del oído en las enfermedades autoinmunes no es infrecuente y en el SS está descrita tanto en la afección primaria como la que se encuentra asociada a otras enfermedades autoinmunes. Cada una de las estructuras anatómicas del oído (interno, medio y externo) puede afectarse. Ello puede traducirse en síntomas como otalgia, acúfenos, vértigo e hipoacusia, y en signos como sequedad de la piel del conducto auditivo, cera seca y ocupación del oído medio por mal funcionamiento de la trompa de Eustaquio (122). La hipoacusia que se puede observar en estos pacientes puede ser neurosensorial, de transmisión o una combinación de ambas. La hipoacusia y los acúfenos pueden aparecer aproximadamente en el 25% de los enfermos, presentándose de forma súbita en algunos de ellos. Hasta la fecha, cinco trabajos han analizado la existencia de hipoacusia neurosensorial en pacientes con SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [23.] Mpl/Fragment 053 01 - Diskussion Bearbeitet: 28. November 2014, 14:43 PlagProf:-) Erstellt: 8. November 2014, 20:44 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 53, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 58, 59, Zeilen: 58: 14 ss. ; 59: 1 ss. |
|---|---|
| [De los 154 pacientes estudiados en total, se ha] detectado hipoacusia en 34 (22%), aunque los porcentajes oscilan de forma importante entre el 5% y el 46%. Respecto a los parámetros inmunológicos, Tumiatti et al (123) han observado una mayor presencia de anticuerpos anticardiolipina (aCL), aunque dicha asociación no ha sido corroborada por trabajos más recientes (124).
En el área nasosinusal la clínica principal es la sensación de sequedad y la formación de costras de moco que padece aproximadamente la sexta parte de los pacientes. La sequedad y atrofia de la mucosa nasal puede explicar la frecuente aparición de epistaxis que se presenta, según las series, entre el 21 y el 35% de los pacientes diagnosticados con SS. Otra clínica frecuente que se observa en los pacientes con el SS son las alteraciones del gusto y del olfato (122). 1.4. Alteraciones analíticas El SS es una enfermedad autoinmune de evolución crónica caracterizada por una respuesta inflamatoria progresiva en las glándulas salivales y lacrimales, que puede afectar de igual manera a órganos extraglandulares. Al igual que en otras enfermedades autoinmunes sistémicas, las alteraciones hematológicas no son raras dentro del SS, e incluso pueden ser la primera manifestación en algunos pacientes. Por otra parte, la determinación sérica de diversas proteínas circulantes puede ser importante en el seguimiento clínico de estos pacientes, y datos analíticos como la hipergammaglobulinemia policlonal o la existencia de una velocidad de sedimentación globular (VSG) elevada, pueden orientar al diagnóstico de SS. |
De los 154 pacientes estudiados en total, se ha detectado hipoacusia en 34 (22%), aunque los porcentajes oscilan de forma importante entre el 5% y el 46%. Respecto a los parámetros inmunológicos, Tumiatti et al (123) han observado una mayor presencia de anticuerpos anticardiolipina (aCL), aunque dicha asociación no ha sido corroborada por trabajos más recientes (124).
En el área nasosinusal la clínica principal es la sensación de sequedad y la formación de costras de moco que padece aproximadamente la sexta parte de los pacientes. La sequedad y atrofia de la mucosa nasal puede explicar la frecuente aparición de epistaxis que se presenta, según las series, entre el 21 y el 35% de los pacientes diagnosticados con SS. Otra clínica frecuente que se observa en los pacientes con el SS son las alteraciones del gusto y del olfato (122). [página 59] 1.4. Alteraciones Analíticas El SS es una enfermedad autoinmune de evolución crónica caracterizada por una respuesta inflamatoria progresiva en las glándulas salivales y lacrimales, que puede afectar de igual manera a órganos extraglandulares. Al igual que en otras enfermedades autoinmunes sistémicas, las alteraciones hematológicas no son raras dentro del SS, e incluso pueden ser la primera manifestación en algunos pacientes. Por otra parte, la determinación sérica de diversas proteínas circulantes puede ser importante en el seguimiento clínico de estos pacientes, y datos analíticos como la hipergammaglobulinemia policlonal o la existencia de una velocidad de sedimentación globular (VSG) elevada, pueden orientar al diagnóstico de SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [24.] Mpl/Fragment 054 01 - Diskussion Bearbeitet: 28. November 2014, 14:47 PlagProf:-) Erstellt: 8. November 2014, 20:46 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 54, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 59, 60, Zeilen: 59: 12 ss. ; 60: 1 ss. |
|---|---|
| 1.4.1. Alteraciones de las proteínas séricas
Las anormalidades en la concentración de las proteínas séricas contribuyen al igual que las alteraciones hematológicas, al gran espectro de manifestaciones biológicas que conforman el SS. a) Reactantes de fase aguda La VSG suele estar elevada en la mayoría de los pacientes con SS e incluso puede sobrepasar los 50 mm en la primera hora en más del 60% de los pacientes (125). En muchas ocasiones, una VSG elevada se relaciona de forma directa con una elevación importante de las proteínas circulantes, especialmente a expensas de una marcada hipergammaglobulinemia. Se han encontrado elevaciones mayores de la VSG en aquellos pacientes con anticuerpos Ro/La (+) (126), describiéndose como un factor predictivo de evolución a SSp en pacientes con síndrome seco (127). La cuantificación de las proteínas totales puede ser superior a 75 g/l en un 80% de los casos y en el proteinograma, las fracciones α1 y α2 pueden estar elevadas en un 20% de los casos (125). Finalmente, no se han realizado estudios sobre el papel diagnóstico de la PCR en el SS, aunque en un estudio preliminar (128) se han descrito valores normales en 22 de 24 pacientes con SSp respecto a pacientes con SS asociado a AR. Por otra parte, Davidson et al (126) no han encontrado diferencias significativas respecto a el valor medio de la PCR en pacientes con SSp agrupados según su perfil inmunológico (seronegativo, ANA/FR o Ro/La). Es posible que la principal utilidad de la PCR en el SSp sea el diagnóstico diferencial entre la infección y la actividad sistémica de la enfermedad. |
1.4.1. Alteraciones de las proteínas séricas
Las anormalidades en la concentración de las proteínas séricas contribuyen al igual que las alteraciones hematológicas, al gran espectro de manifestaciones biológicas que conforman el SS. a) Reactantes de fase aguda La VSG suele estar elevada en la mayoría de los pacientes con SS e incluso puede sobrepasar los 50 mm en la primera hora en más del 60% de los pacientes (125). En muchas ocasiones, una VSG elevada se relaciona de forma directa con una elevación importante de las proteínas circulantes, especialmente a expensas de una marcada hipergammaglobulinemia. Se han encontrado elevaciones mayores de la VSG en aquellos pacientes con [página 60] anticuerpos Ro/La (+) (126), describiéndose como un factor predictivo de evolución a SSp en pacientes con síndrome seco (127). La cuantificación de las proteínas totales puede ser superior a 75 g/l en un 80% de los casos y en el proteinograma, las fracciones α1 y α2 pueden estar elevadas en un 20% de los casos (125). Finalmente, no se han realizado estudios sobre el papel diagnóstico de la PCR en el SS, aunque en un estudio preliminar (128) se han descrito valores normales en 22 de 24 pacientes con SSp respecto a pacientes con SS asociado a AR. Por otra parte, Davidsson et al (126) no han encontrado diferencias significativas respecto a el valor medio de la PCR en pacientes con SSp agrupados según su perfil inmunológico (seronegativo, ANA/FR o Ro/La). Es posible que la principal utilidad de la PCR en el SSp sea el diagnóstico diferencial entre la infección y la actividad sistémica de la enfermedad. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [25.] Mpl/Fragment 055 01 - Diskussion Bearbeitet: 28. November 2014, 14:50 PlagProf:-) Erstellt: 8. November 2014, 22:43 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 55, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 60, 61, Zeilen: 60: 14 ss.;61: 1 ss. |
|---|---|
| 1.4.2. Alteraciones de las gammaglobulinas
La elevación de las gammaglobulinas séricas es uno de los principales datos analíticos característicos del SS, con una prevalencia que puede alcanzar hasta el 70% de los casos (125). Otros tipos de alteraciones descritos son la aparición de una banda monoclonal y la existencia de hipogammaglobulinemia o déficits de ciertas inmunoglobulinas. a) Hipergammaglobulinemia policlonal La hipergammaglobulinemia policlonal es uno de los datos analíticos más característicos del SSp y es el reflejo directo de la hiperactividad linfocitaria característica de la enfermedad. Aunque se ha descrito tanto en el SSp como en el secundario, ya en los estudios clásicos de los años 70 se consideró como un dato más representativo de la forma primaria de la enfermedad. Aunque en los estudios clásicos (12,129) se describe la existencia de hipergammaglobulinemia en casi el 100% de los pacientes con SS, estudios posteriores describen porcentajes inferiores que oscilan entre el 36-42% (12,129-132) (Tabla 3). TABLA 3. Prevalencia de hipergammaglobulinemia policlonal en el SSp
NE, no especificado |
1.4.2. Alteraciones de las gammaglobulinas
La elevación de las gammaglobulinas séricas es uno de los principales datos analíticos característicos del SS, con una prevalencia que puede alcanzar hasta el 70% de los casos (125). Otros tipos de alteraciones descritos son la aparición de una banda monoclonal y la existencia de hipogammaglobulinemia o déficits de ciertas inmunoglobulinas. a) Hipergammaglobulinemia policlonal La hipergammaglobulinemia policlonal es uno de los datos analíticos más característicos del SSp y es el reflejo directo de la hiperactividad linfocitaria característica de la enfermedad. Aunque se ha descrito tanto en el [página 61] SSp como en el secundario, ya en los estudios clásicos de los años 70 se consideró como un dato más representativo de la forma primaria de la enfermedad. Aunque en los estudios clásicos (12,129) se describe la existencia de hipergammaglobulinemia en casi el 100% de los pacientes con SS, estudios posteriores describen porcentajes inferiores que oscilan entre el 36-42% (12,129-132) (Tabla 3). TABLA 3. Prevalencia de hipergammaglobulinemia policlonal en el SSp
NE, no especificado |


Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [26.] Mpl/Fragment 056 01 - Diskussion Bearbeitet: 28. November 2014, 14:53 PlagProf:-) Erstellt: 8. November 2014, 22:45 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 56, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 61, 62, Zeilen: 61: 8 ss.; 62: 1 ss. |
|---|---|
| Alexander et al (130) encontraron cifras de gammaglobulinas >4g/l en el 36% de pacientes con SS, mientras que en el estudio reciente de Skopouli et al (132) se encontró una hipergammaglobulinemia (>2g/l) en el 42% de los pacientes con SS.
Los trastornos ocasionados por la elevación de las gammaglobulinas son excepcionales y no suelen observarse fenómenos clínicos de hiperviscosidad ni defectos en la acidificación renal causados por la hipergammaglobulinemia, aunque Skopouli et al (132), describieron un paciente con una importante hipergammaglobulinemia que murió por un tromboembolismo pulmonar severo. En cambio, la relación entre la hipergammaglobulinemia y la afección cutánea en forma de púrpura fue descrita en pacientes con SS en los años 60 y 70, bajo el nombre de púrpura hiperglobulinémica de Waldenström, considerándose incluso como una entidad independiente (133,134). Posteriormente se ha comprobado que en los pacientes con SSp, la existencia de púrpura e hipergammaglobulinemia probablemente forman parte del conjunto de manifestaciones extraglandulares y biológicas asociadas al SSp. Finalmente, la existencia de hipergammaglobulinemia se ha asociado con la positividad de los anticuerpos anti-Ro/SSA. Alexander et al (130) encontraron que los pacientes que muestran positividad ante dichos anticuerpos son serológicamente más reactivos que aquellos que no los presentan. Respecto a la elevación específica de las subclases de inmunoglobulinas, lo más frecuente es el aumento de la IgG >1.5gr/L, que se ha reportado en el 80% de los casos (125) y algún estudio ha demostrado que los pacientes con síndrome seco y niveles elevados de IgG, acaban por desarrollar con más frecuencia un SSp (127). Los niveles de IgA suelen ser normales, aunque los de IgA secretora suelen ser el doble de lo normal (125). |
Alexander et al (130) encontraron cifras de gammaglobulinas >4g/l en el 36% de pacientes con SS, mientras que en el estudio reciente de Skopouli et al (132) se encontró una hipergammaglobulinemia (>2g/l) en el 42% de los pacientes con SS.
Los trastornos ocasionados por la elevación de las gammaglobulinas son excepcionales y no suelen observarse fenómenos clínicos de hiperviscosidad ni [página 62] defectos en la acidificación renal causados por la hipergammaglobulinemia, aunque Skopouli et al (132), describieron un paciente con una importante hipergammaglobulinemia que murió por un tromboembolismo pulmonar severo. En cambio, la relación entre la hipergammaglobulinemia y la afección cutánea en forma de púrpura fue descrita en pacientes con SS en los años 60 y 70, bajo el nombre de púrpura hiperglobulinémica de Waldenström, considerándose incluso como una entidad independiente (133,134). Posteriormente se ha comprobado que en los pacientes con SSp, la existencia de púrpura e hipergammaglobulinemia probablemente forman parte del conjunto de manifestaciones extraglandulares y biológicas asociadas al SSp. Finalmente, la existencia de hipergammaglobulinemia se ha asociado con la positividad de los anticuerpos anti- Ro/SSA. Alexander et al (130) encontraron que los pacientes que muestran positividad ante dichos anticuerpos son serológicamente más reactivos que aquellos que no los presentan. Respecto a la elevación específica de las subclases de inmunoglobulinas, lo más frecuente es el aumento de la IgG >1.5gr/L, que se ha reportado en el 80% de los casos (125) y algún estudio ha demostrado que los pacientes con síndrome seco y niveles elevados de IgG, acaban por desarrollar con más frecuencia un SSp (127). Los niveles de IgA suelen ser normales, aunque los de IgA secretora suelen ser el doble de lo normal (125). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [27.] Mpl/Fragment 057 01 - Diskussion Bearbeitet: 28. November 2014, 14:56 PlagProf:-) Erstellt: 8. November 2014, 22:48 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 57, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 62, 63, 64, Zeilen: 62: 21 ss. ; 63: 1 ss.; 64: 1 ss. |
|---|---|
| [Los niveles de IgM generalmente son normales (125), aunque se han] observado elevaciones policlonales en pacientes con SS asociado a esclerodermia (ES) o a la presencia de niveles elevados de factor reumatoide (FR).
b) Hipogammaglobulinemia y déficits de inmunoglobulinas La presencia de hipogammaglobulinemia en pacientes con SS se ha considerado clásicamente como presagio de linfoma maligno. Bloch et al (12) encontraron una prevalencia de hipogammaglobulinemia del 3% (2 de 62 pacientes), los 2 casos asociados a reticulosarcomas. Por otra parte, estudios recientes han encontrado que cerca de un 20% de pacientes con SSp presentan cifras bajas (<1.5g/l) de gammaglobulinas, un dato analítico asociado a una menor frecuencia de anticuerpos anti-Ro y anti-La (135). Por otra parte, se han descrito algunos casos de deficiencias de inmunoglobulinas asociadas al SSp. La inmunodeficiencia descrita con más frecuencia en el SS es el déficit de IgA, aunque debe considerarse como una asociación muy infrecuente ya que desde 1971 sólo se han reportado 7 casos (136-142) (Tabla 4). En los 7 pacientes descritos con SS y deficiencia selectiva de IgA, sólo se han descrito infecciones de repetición en un paciente y episodios de parotiditis en otros 2. Otro trastorno menos frecuente es la deficiencia de IgG2, que se ha relacionado con una mayor susceptibilidad a infecciones bacterianas neumocócicas (139). Sin embargo, Eriksson et al (141) mencionan que los pacientes con SS pueden tener una deficiencia de IgG2 a pesar de tener niveles elevados de IgG, y que tal deficiencia no predispone por sí sola a desarrollar infecciones. |
Los niveles de IgM generalmente son normales (125), aunque se han observado elevaciones policlonales en pacientes con SS asociado a esclerodermia (ES) o a la presencia de niveles elevados de factor reumatoide (FR).
[página 63] b) Hipogammaglobulinemia y déficits de inmunoglobulinas La presencia de hipogammaglobulinemia en pacientes con SS se ha considerado clásicamente como presagio de linfoma maligno. Bloch et al (12) encontraron una prevalencia de hipogammaglobulinemia del 3% (2 de 62 pacientes), los 2 casos asociados a reticulosarcomas. Por otra parte, estudios recientes han encontrado que cerca de un 20% de pacientes con SSp presentan cifras bajas (<1.5g/l) de gammaglobulinas, un dato analítico asociado a una menor frecuencia de anticuerpos anti-Ro y anti-La (135). Por otra parte, se han descrito algunos casos de deficiencias de inmunoglobulinas asociadas al SSp. La inmunodeficiencia descrita con más frecuencia en el SS es el déficit de IgA, aunque debe considerarse como una asociación muy infrecuente ya que desde 1971 sólo se han reportado 7 casos (136-142) (Tabla 4). [...] [página 64] En los 7 pacientes descritos con SS y deficiencia selectiva de IgA, sólo se han descrito infecciones de repetición en un paciente y episodios de parotiditis en otros 2. Otro trastorno menos frecuente es la deficiencia de IgG2, que se ha relacionado con una mayor susceptibilidad a infecciones bacterianas neumocócicas (139). Sin embargo, Eriksson et al (141) mencionan que los pacientes con SS pueden tener una deficiencia de IgG2 a pesar de tener niveles elevados de IgG, y que tal deficiencia no predispone por sí sola a desarrollar infecciones. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [28.] Mpl/Fragment 058 01 - Diskussion Bearbeitet: 28. November 2014, 15:00 PlagProf:-) Erstellt: 8. November 2014, 23:19 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 58, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 63, 64, 65, Zeilen: 63: tabla; 64: 10 ss. ; 65: 1 ss. |
|---|---|
| TABLA 4. Deficiencias de inmunoglobulinas en pacientes con SSp
c) Inmunoglobulinas monoclonales circulantes La existencia de inmunoglobulinas monoclonales circulantes es un dato frecuente en pacientes con SS, ya que se piensa que las inmunoglobulinas monoclonales y el FR se producen en etapas tempranas de forma local en las glándulas salivales de estos pacientes. Aunque la aparición de un pico monoclonal cuantitativamente importante puede ser la primera manifestación biológica del desarrollo de un proceso linfoproliferativo, no es infrecuente observar en pacientes con SSp, la existencia de una gammapatía monoclonal de significado incierto (GMSI) que obliga a descartar su asociación con un síndrome crioglobulinémico y a realizar en estos pacientes un estrecho seguimiento clínico (144). Mediante electroforesis en gel de agarosa de alta resolución, capaz de detectar cantidades mínimas de gamaglobulinas indetectables por métodos electroforéticos de rutina, se pueden detectar cadenas monoclonales en el 47% de los pacientes en suero y en el 76% en orina (125). En un estudio de 1983, se observó la presencia de cadenas ligeras monoclonales en el 100% de los pacientes con SS que presentaban manifestaciones extraglandulares frente al 22% de pacientes que [presentaban exclusivamente un síndrome seco (145).] |
TABLA 4. Deficiencias de inmunoglobulinas en pacientes con SSp
[página 64] c) Inmunoglobulinas monoclonales circulantes La existencia de inmunoglobulinas monoclonales circulantes es un dato frecuente en pacientes con SS, ya que se piensa que las inmunoglobulinas monoclonales y el FR se producen en etapas tempranas de forma local en las glándulas salivales de estos pacientes. Aunque la aparición de un pico monoclonal cuantitativamente importante puede ser la primera manifestación biológica del desarrollo de un proceso linfoproliferativo, no es infrecuente observar en pacientes con SSp, la existencia de una gammapatía monoclonal de significado incierto (GMSI) que obliga a descartar su asociación con un síndrome crioglobulinémico y a realizar en estos pacientes un estrecho seguimiento clínico (144). Mediante electroforesis en gel de agarosa de alta resolución, capaz de detectar cantidades mínimas de gamaglobulinas indetectables por métodos electroforéticos de rutina, se pueden detectar cadenas monoclonales en el 47% de los pacientes en suero y en el 76% en orina (125). En un estudio de 1983, se observó la presencia de cadenas ligeras [página 65] monoclonales en el 100% de los pacientes con SS que presentaban manifestaciones extraglandulares frente al 22% de pacientes que presentaban exclusivamente un síndrome seco (145). |
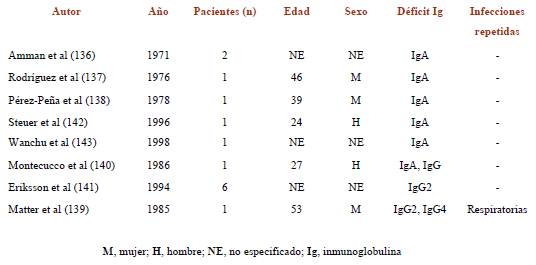

Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [29.] Mpl/Fragment 059 01 - Diskussion Bearbeitet: 28. November 2014, 15:04 PlagProf:-) Erstellt: 8. November 2014, 23:21 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 59, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 65, 66, Zeilen: 65: 3 ss.; 66: 1 ss. |
|---|---|
| En un estudio japonés (146) se describió la presencia de cadenas ligeras monoclonales en el 70% de los pacientes con SS y manifestaciones extraglandulares, además de encontrar bandas ligeras en orina. En este estudio, se describieron 18 pacientes con SS e inmunoglobulinas monoclonales: 8 bandas de tipo IgA, 4 IgG, 4 IgM y 2 con doble banda monoclonal (IgMκ/IgGκ e IgAκ/IgGκ). Por otra parte, se revela la importancia de la GMSI como complicación del SS en un grupo de pacientes japoneses y no japoneses. En ambos grupos, las inmunoglobulinas monoclonales predominantes fueron: 27 IgM, 11 IgA, 6 IgG y una proteína de Bence Jones en un paciente japonés (146).
d) β2 microglobulina La β2 microglobulina es una proteína de bajo peso molecular (11.700 daltons) secretada por células nucleadas (linfocitos y otras células); forma la cadena ligera de los antígenos leucocitarios humanos (HLA) y se une de forma no covalente a diversas glucoproteínas transmembrana, como a la molécula HLA clase I. Normalmente se encuentra en concentraciones bajas en suero, líquidos corporales y secreciones (147). Los niveles de β2 microglobulina en suero se encuentran elevados en pacientes con SS y esta elevación se ha relacionado con algunos aloantígenos de histocompatibilidad. En un estudio realizado en 24 pacientes con SS se encontró que en aquellos con elevación de la β2 microglobulina predominaba el haplotipo HLA DR3 y en menor frecuencia el B8 (148). Por otra parte, los niveles de β2 microglobulina se han considerado como un factor predictivo de evolución a SSp en pacientes con síndrome seco (127). También se han detectado niveles elevados de β2 microglobulina en las glándulas salivales de pacientes con SSp, indicando de esta manera su relación con el [grado de infiltración linfocitaria.] |
En un estudio japonés (146) se describió la presencia de cadenas ligeras monoclonales en el 70% de los pacientes con SS y manifestaciones extraglandulares, además de encontrar bandas ligeras en orina. En este estudio, se describieron 18 pacientes con SS e inmunoglobulinas monoclonales: 8 bandas de tipo IgA, 4 IgG, 4 IgM y 2 con doble banda monoclonal (IgMκ/IgGκ e IgAκ/IgGκ). Por otra parte, se revela la importancia de la GMSI como complicación del SS en un grupo de pacientes japoneses y no japoneses. En ambos grupos, las inmunoglobulinas monoclonales predominantes fueron: 27 IgM, 11 IgA, 6 IgG y una proteína de Bence Jones en un paciente japonés (146).
d) β2 microglobulina La β2 microglobulina es una proteína de bajo peso molecular (11.700 daltons) secretada por células nucleadas (linfocitos y otras células); forma la cadena ligera de los antígenos leucocitarios humanos (HLA) y se une de forma no covalente a diversas glucoproteínas transmembrana, como a la molécula HLA clase I. Normalmente se encuentra en concentraciones bajas en suero, líquidos corporales y secreciones (147). Los niveles de β2 microglobulina en suero se encuentran elevados en pacientes con SS y esta elevación se ha relacionado con algunos aloantígenos de histocompatibilidad. En un estudio realizado en 24 pacientes con SS se encontró que en aquellos con elevación de la β2 microglobulina predominaba el haplotipo HLA DR3 y en menor frecuencia el B8 (148). Por otra parte, los [página 66] niveles de β2 microglobulina se han considerado como un factor predictivo de evolución a SSp en pacientes con síndrome seco (127). También se han detectado niveles elevados de β2 microglobulina en las glándulas salivales de pacientes con SSp, indicando de esta manera su relación con el grado de infiltración linfocitaria. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [30.] Mpl/Fragment 060 01 - Diskussion Bearbeitet: 28. November 2014, 15:07 PlagProf:-) Erstellt: 8. November 2014, 23:28 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 60, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 66, 67, Zeilen: 66: 5 ss.; 67: 1 ss. |
|---|---|
| En un estudio realizado en 49 pacientes con sospecha de SS se encontró una concentración elevada en saliva de la β2 microglobulina (>4 μg/ml) en 35% de ellos, todos diagnosticados de SSp, mientras que el 65% restante, presentaron un SS asociado o bien un síndrome seco (147).
Además de la elevada concentración de la β2 microglobulina presente en las glándulas salivales de pacientes afectos con SSp, también se han encontrado niveles significativos de esta proteína en suero de pacientes con diversas afectaciones extraglandulares, especialmente a nivel pulmonar, nefrológico y linfoproliferativo. Se ha propuesto que concentraciones elevadas de la β2 microglobulina en pacientes con enfisema pulmonar se asocian con la existencia de una enfermedad obstructiva de las vías aéreas pequeñas, siendo los niveles de esta proteína en suero inversamente proporcionales a la capacidad vital forzada (CVF), volumen espiratorio forzado en el primer segundo (FEV1) y la DLCO (149). También se han descrito niveles elevados de β2 microglobulina en pacientes con SSp y alveolitis linfocítica o neutrofílica (62). Por otro lado, se ha estudiado la relación entre niveles elevados de β2- microglobulina y la afección renal en el SS. Pertovaara et al (150) encontraron ATRd en 18 de 55 pacientes (33%), los cuales mostraron niveles elevados de β2 microglobulina en suero respecto a los pacientes con capacidad de acidificación normal. Esto sugiere que los niveles elevados de β2 microglobulina en pacientes con afección renal, traducen la existencia de una prolongada activación linfocitaria y por lo tanto, reflejan la severidad del SS. También se observó una correlación entre la presencia de niveles elevados de β2 microglobulina con la existencia de hipertensión y una duración prolongada de los síntomas de sequedad. Finalmente, también se ha relacionado la existencia de niveles elevados de la β2 microglobulina con la presencia de tumores [malignos.] |
En un estudio realizado en 49 pacientes con sospecha de SS se encontró una concentración elevada en saliva de la β2 microglobulina (>4 μg/ml) en 35% de ellos, todos diagnosticados de SSp, mientras que el 65% restante, presentaron un SS asociado o bien un síndrome seco (147).
Además de la elevada concentración de la β2 microglobulina presente en las glándulas salivales de pacientes afectos con SSp, también se han encontrado niveles significativos de esta proteína en suero de pacientes con diversas afectaciones extraglandulares, especialmente a nivel pulmonar, nefrológico y linfoproliferativo. Se ha propuesto que concentraciones elevadas de la β2 microglobulina en pacientes con enfisema pulmonar se asocian con la existencia de una enfermedad obstructiva de las vías aéreas pequeñas, siendo los niveles de esta proteína en suero inversamente proporcionales a la capacidad vital forzada (CVF), volumen espiratorio forzado en el primer segundo (FEV1) y la DLCO (149). También se han descrito niveles elevados de β2 microglobulina en pacientes con SSp y alveolitis linfocítica o neutrofílica (62). Por otro lado, se ha estudiado la relación entre niveles elevados de β2- microglobulina y la afección renal en el SS. Pertovaara et al (150) encontraron ATRdl en 18 de 55 pacientes (33%), los cuales mostraron niveles elevados de β2 microglobulina en suero respecto a los pacientes con capacidad de [página 67] acidificación normal. Esto sugiere que los niveles elevados de β2 microglobulina en pacientes con afección renal, traducen la existencia de una prolongada activación linfocitaria y por lo tanto, reflejan la severidad del SS. También se observó una correlación entre la presencia de niveles elevados de β2 microglobulina con la existencia de hipertensión y una duración prolongada de los síntomas de sequedad. Finalmente, también se ha relacionado la existencia de niveles elevados de la β2 microglobulina con la presencia de tumores malignos. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [31.] Mpl/Fragment 061 01 - Diskussion Bearbeitet: 28. November 2014, 15:55 PlagProf:-) Erstellt: 9. November 2014, 00:12 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 61, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 67, 68, Zeilen: 67: 8 ss.; 68: 1 ss. |
|---|---|
| Michalski et al (147) encontraron una elevación de la β2 microglobulina en el 77% de pacientes con SS asociado a linfoma o pseudolinfoma.
De acuerdo con lo descrito anteriormente, la detección de la β2 microglobulina en saliva y suero puede ser un método útil y sencillo para estimar el grado de infiltración linfocitaria activa en los órganos afectos por el SS. También se podría utilizar como un parámetro útil de laboratorio en respuesta al tratamiento, ya que el incremento en proliferación y aumento de masa linfoidea se correlaciona con los niveles de β2 microglobulina (147). 1.4.3. Alteraciones hematológicas La existencia de alteraciones hematológicas es un dato analítico que con frecuencia nos encontramos en pacientes con SSp. Diversos autores han descrito alteraciones analíticas en el 40 y el 50% de los pacientes con SSp. Aunque en la mayoría de las ocasiones dichas alteraciones no poseen una traducción clínica, se han descrito diversos casos de debut de un SSp por una hemocitopenia grave y sintomática. a) Alteraciones en la serie roja La anemia está presente entre el 16 y el 50% (según las distintas series) de los pacientes con SS (12,129,130,132,151) (Tabla 5). TABLA 5. Prevalencia de anemia en pacientes con SS
|
Michalsky et al (147), encontraron una elevación de la β2 microglobulina en el 77% de pacientes con SS asociado a linfoma o pseudolinfoma.
De acuerdo con lo descrito anteriormente, la detección de la β2 microglobulina en saliva y suero puede ser un método útil y sencillo para estimar el grado de infiltración linfocitaria activa en los órganos afectos por el SS. También se podría utilizar como un parámetro útil de laboratorio en respuesta al tratamiento, ya que el incremento en proliferación y aumento de masa linfoidea se correlaciona con los niveles de β2 microglobulina (147). 1.4.3. Alteraciones hematológicas La existencia de alteraciones hematológicas es un dato analítico que con frecuencia nos encontramos en pacientes con SSp. Diversos autores han descrito alteraciones analíticas en el 40 y el 50% de los pacientes con SSp. Aunque en la mayoría de las ocasiones dichas alteraciones no poseen una traducción clínica, se han descrito diversos casos de debut de un SSp por una hemocitopenia grave y sintomática. [página 68] a) Alteraciones en la serie roja La anemia está presente entre el 16 y el 50% (según las distintas series) de los pacientes con SS (12,129,130,132,151) (Tabla 5). TABLA 5. Prevalencia de anemia en pacientes con SS
|

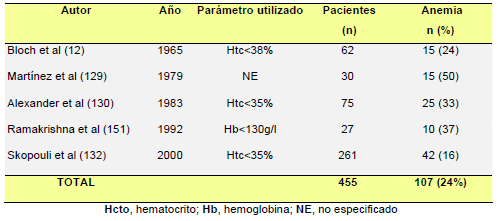
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [32.] Mpl/Fragment 062 01 - Diskussion Bearbeitet: 28. November 2014, 17:43 Singulus Erstellt: 9. November 2014, 00:22 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 62, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 68, 69, Zeilen: 68: 4 ss; 69: 1 ss. |
|---|---|
| En general, el tipo más frecuente de anemia es similar a la que se observa en las enfermedades inflamatorias crónicas, es decir, normocítica y normocrómica (125,129). Cabe mencionar que en ocasiones la anemia puede ser debida a causas farmacológicas (125) por ejemplo, a tratamientos prolongados con dosis altas de antiinflamatorios no esteroideos (AINES) (hemorragia digestiva) y corticoides (hipoplasia medular e insuficiencia renal).
Existen pocos estudios que hayan analizado las características de la anemia en el SS. En 1965, Bloch et al (12), encontraron anemia en 15/62 (24%) pacientes, aunque solamente 3 tenían un Hcto <30% (2 por una anemia ferropénica y 1 por un síndrome de Felty, en el cual la anemia revirtió tras la realización de una esplenectomía). Alexander et al (130), en un estudio realizado en 75 pacientes, encontraron anemia en un 33% de pacientes siendo la mayoría de ellos anti-Ro/SSA (+). El anticuerpo anti-Ro/SSA puede coexistir con marcadores de hiperrreactividad de células B (hipergammaglobulinemia, FR) y otras citopenias apoyando así una patogénesis autoinmune de la anemia (152). Ramakrishna et al (151) encontraron anemia en 10 de 27 pacientes (37%), aunque sólo describieron una causa evidente de anemia en 7 de ellos: 3 con anemia hemolítica autoinmune (AHAI), 2 con anemia refractaria asociada a síndrome mielodisplásico, 1 con aplasia pura de la serie eritrocitaria y otro paciente con anemia aplásica. Realmente, la mayoría de estos procesos hematológicos suelen ser una causa excepcional de anemia en el SSp que a continuación analizaremos por separado (130,132,141,151-163) (Tabla 6). |
En general, el tipo más frecuente de anemia es similar a la que se observa en las enfermedades inflamatorias crónicas, es decir, normocítica y normocrómica (125,129). Cabe mencionar que en ocasiones la anemia puede ser debida a causas farmacológicas (125) por ejemplo, a tratamientos prolongados con dosis altas de antiinflamatorios no esteroideos (AINES) (hemorragia digestiva) y corticoides (hipoplasia medular e insuficiencia renal).
Existen pocos estudios que hayan analizado las características de la anemia en el SS. En 1965, Bloch et al (12), encontraron anemia en 15/62 (24%) pacientes, aunque solamente 3 tenían un Hcto <30% (2 por una anemia ferropénica y 1 por un síndrome de Felty, en el cual la anemia revirtió tras la realización de una esplenectomía). Alexander et al (130), en un estudio realizado en 75 pacientes, encontraron anemia en un 33% de pacientes siendo la mayoría de ellos anti-Ro/SSA (+). El anticuerpo anti-Ro/SSA puede coexistir [página 69] con marcadores de hiperrreactividad de células B (hipergammaglobulinemia, FR) y otras citopenias apoyando así una patogénesis autoinmune de la anemia (152). Ramakrishna et al (151) encontraron anemia en 10 de 27 pacientes (37%), aunque sólo describieron una causa evidente de anemia en 7 de ellos: 3 con anemia hemolítica autoinmune (AHAI), 2 con anemia refractaria asociada a síndrome mielodisplásico, 1 con aplasia pura de la serie eritrocitaria y otro paciente con anemia aplásica. Realmente, la mayoría de estos procesos hematológicos suelen ser una causa excepcional de anemia en el SSp que a continuación analizaremos por separado (130,132,141,151-163) (Tabla 6). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [33.] Mpl/Fragment 063 01 - Diskussion Bearbeitet: 28. November 2014, 17:49 Singulus Erstellt: 9. November 2014, 00:45 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 63, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 69, 70, Zeilen: 79: tabla; 70: 1 ss. |
|---|---|
| TABLA 6. Pacientes con SSp y causas infrecuentes de anemia
Anemia hemolítica autoinmune. Aunque algunos estudios consideran la positividad a la prueba de Coombs, como una alteración hematológica frecuente en el SS, entre el 22 al 47% de los pacientes, la hemólisis franca es poco habitual (152). La AHAI es una manifestación hematológica infrecuente del SSp, ya que hasta la fecha sólo se han descrito 22 pacientes (Tabla 6). Sin embargo, suele hallarse con más frecuencia en pacientes con SS asociado (asociado a AR, LES o ES) o asociado a procesos linfoproliferativos malignos (154). De los casos con SS y AHAI publicados, la mayoría fueron tratados con corticoides, obteniendo buena respuesta, aunque en 3 [pacientes fue necesario añadir un tratamiento inmunosupresor (azatioprina en 2 y ciclofosfamida en 1 caso) (140,151,152,154,155,157) (Tabla 7).] |
TABLA 6. Pacientes con SSp y causas infrecuentes de anemia
[página 70] Anemia hemolítica autoinmune. Aunque algunos estudios consideran la positividad a la prueba de Coombs, como una alteración hematológica frecuente en el SS, entre el 22 al 47% de los pacientes, la hemólisis franca es poco habitual (152). La AHAI es una manifestación hematológica infrecuente del SSp, ya que hasta la fecha sólo se han descrito 22 pacientes (Tabla 6). Sin embargo, suele hallarse con más frecuencia en pacientes con SSs (asociado a AR, LES o ES) o asociado a procesos linfoproliferativos malignos (154). De los casos con SS y AHAI publicados, la mayoría fueron tratado [sic] con corticoides, obteniendo buena respuesta, aunque en 3 pacientes fue necesario añadir un tratamiento inmunosupresor (azatioprina en 2 y ciclofosfamida en 1 caso) (140,151,152,154,155,157) (Tabla 7). |
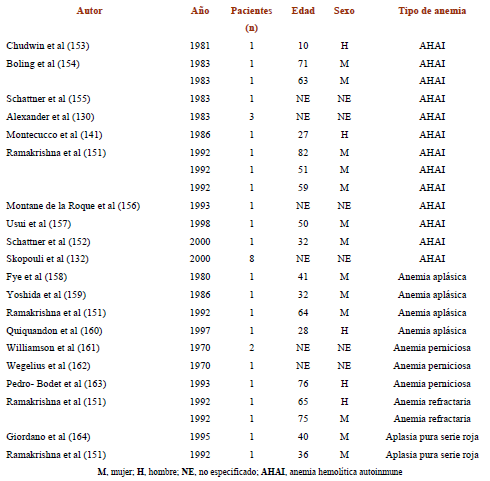
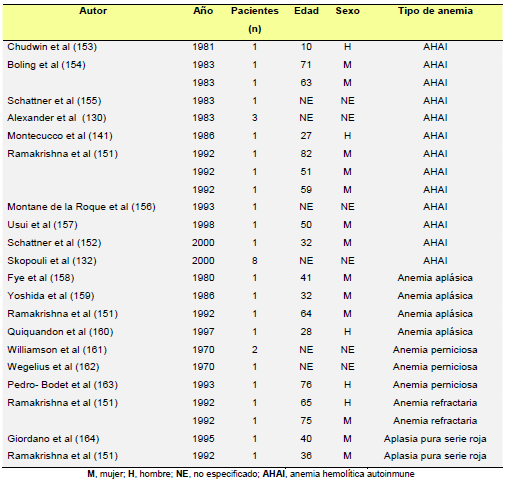
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [34.] Mpl/Fragment 064 01 - Diskussion Bearbeitet: 28. November 2014, 18:00 Hindemith Erstellt: 9. November 2014, 00:49 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 64, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 70, 71, Zeilen: 70: 8 ss.; 71: 1 ss. |
|---|---|
| [De los casos con SS y AHAI publicados, la mayoría fueron tratados con corticoides, obteniendo buena respuesta, aunque en 3] pacientes fue necesario añadir un tratamiento inmunosupresor (azatioprina en 2 y ciclofosfamida en 1 caso) (140,151,152,154,155,157) (Tabla 7).
TABLA 7. Pacientes con SSp y AHAI: tratamientos recibidos y evolución
De forma excepcional, la AHAI puede ser la primera manifestación hematológica de un SSp incipiente (152), por ello, dada la variabilidad de presentación del SS, es imprescindible incluir al SSp en el diagnóstico diferencial de la AHAI. La causa de la anemia hemolítica en el SS se desconoce. La hiperactividad de los linfocitos B con la consiguiente producción de múltiples autoanticuerpos es el dato característico del SS y no cabría descartar la posible producción de anticuerpos antieritrocitarios como causantes de la anemia hemolítica en esta enfermedad (154). Es de importancia mencionar la relación que se ha encontrado entre la anemia hemolítica y la presencia de anticuerpos anti-Ro/SSA. El mecanismo por el cual los anticuerpos anti-Ro/SSA y anti-La/SSB puedan intervenir directamente en el desarrollo de citopenias, se relacionaría con la existencia de diversos estímulos que provocarían la expresión anómala de los antígenos SSA y SSB en la membrana celular cuando habitualmente se localizan en el núcleo (151). |
De los casos con SS y AHAI publicados, la mayoría fueron tratado [sic] con corticoides, obteniendo buena respuesta, aunque en 3 pacientes fue necesario añadir un tratamiento inmunosupresor (azatioprina en 2 y ciclofosfamida en 1 caso) (140,151,152,154,155,157) (Tabla 7).
TABLA 7. Pacientes con SSp y AHAI: tratamientos recibidos y evolución
De forma excepcional, la AHAI puede ser la primera manifestación hematológica de un SSp incipiente (152), por ello, dada la variabilidad de [página 71] presentación del SS, es imprescindible incluir al SSp en el diagnóstico diferencial de la AHAI. La causa de la anemia hemolítica en el SS se desconoce. La hiperactividad de los linfocitos B con la consiguiente producción de múltiples autoanticuerpos es el dato característico del SS y no cabría descartar la posible producción de anticuerpos antieritrocitarios como causantes de la anemia hemolítica en esta enfermedad (154). Es de importancia mencionar la relación que se ha encontrado entre la anemia hemolítica y la presencia de anticuerpos anti-Ro/SSA. El mecanismo por el cual los anticuerpos anti-Ro/SSA y anti-La/SSB puedan intervenir directamente en el desarrollo de citopenias, se relacionaría con la existencia de diversos estímulos que provocarían la expresión anómala de los antígenos SSA y SSB en la membrana celular cuando habitualmente se localizan en el núcleo (151). |
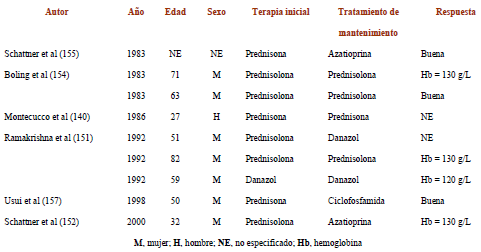
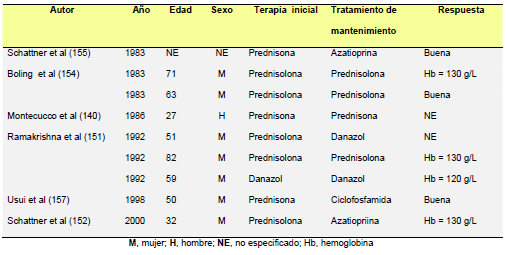
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [35.] Mpl/Fragment 065 01 - Diskussion Bearbeitet: 28. November 2014, 18:01 Hindemith Erstellt: 9. November 2014, 09:59 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 65, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 71, 72, Zeilen: 71: 13 ss.; 72: 1 ss. |
|---|---|
| [Estos estímulos pueden ser tan diversos como la] exposición de las células a rayos ultravioleta o infecciones por diversos virus, como el adenovirus o aquellos con un determinado tropismo medular, que inducirían la lisis por autoanticuerpos de los hematíes. Finalmente, se ha descrito un caso de anemia hemolítica en un recién nacido de una madre con SS portadora de anticuerpo anti- Ro/SSA, lo que muestra la implicación de los anticuerpos anti-Ro/SSA en las alteraciones hematológicas de la enfermedad (151).
Aplasia medular. Aunque el primer caso de anemia aplásica en el SS se describió en un paciente con linfoma, se han publicado 4 casos (151,158-160) (Tabla 6) no asociados a procesos linfoproliferativos (160). La anemia aplásica también se ha descrito asociada a otras enfermedades del tejido conectivo, particularmente con el LES, aunque generalmente se debe al tratamiento inmunodepresor. Hasta la fecha, no se han realizado estudios específicos en pacientes con SS y anemia aplásica para delinear los mecanismos o para estudiar la existencia de inhibidores de formación de colonias en suero (151). Es probable que un mecanismo autoinmune similar al descrito en el LES esté implicado en la patogenia del SS con anemia aplásica. Por otra parte, se ha reportado un paciente con la traslocación (q24, p23) en el gen (14q 24), donde está localizado el gen que codifica el factor de crecimiento tumoral β, una linfocina hematopoyética supresora. Dicha translocación podría conducir a una expresión anormal del gen del factor de crecimiento tumoral β y por lo tanto, a la supresión de la hematopoyesis (160). Estudios in vitro, han demostrado la existencia de anticuerpos IgG (dependientes o no de complemento) que inhiben la proliferación de la médula ósea (151) en pacientes con LES, aunque no se han realizado estudios similares en pacientes con SS. |
Estos estímulos pueden ser tan diversos como la exposición de las células a rayos ultravioleta o infecciones por diversos virus, como el adenovirus o aquellos con un determinado tropismo medular, que inducirían la lisis por autoanticuerpos de los hematíes. Finalmente, se ha descrito un caso de anemia hemolítica en un recién nacido de una madre con SS portadora de anticuerpo anti- Ro/SSA, lo que muestra la implicación de los anticuerpos anti-Ro/SSA en las alteraciones hematológicas de la enfermedad (151).
Aplasia medular. Aunque el primer caso de anemia aplásica en el SS se describió en un paciente con linfoma, se han publicado 4 casos (151,158- 160) (Tabla 6) no asociados a procesos linfoproliferativos (160). La anemia aplásica también se ha descrito asociada a otras enfermedades del tejido [página 72] conectivo, particularmente con el LES, aunque generalmente se debe al tratamiento inmunodepresor. Hasta la fecha, no se han realizado estudios específicos en pacientes con SS y anemia aplásica para delinear los mecanismos o para estudiar la existencia de inhibidores de formación de colonias en suero (151). Es probable que un mecanismo autoinmune similar al descrito en el LES esté implicado en la patogenia del SS con anemia aplásica. Por otra parte, se ha reportado un paciente con la traslocación (q24, p23) en el gen (14q 24), donde está localizado el gen que codifica el factor de crecimiento tumoral β, una linfocina hematopoyética supresora. Dicha translocación podría conducir a una expresión anormal del gen del factor de crecimiento tumoral β y por lo tanto, a la supresión de la hematopoyesis (160). Estudios in vitro, han demostrado la existencia de anticuerpos IgG (dependientes o no de complemento) que inhiben la proliferación de la médula ósea (151) en pacientes con LES, aunque no se han realizado estudios similares en pacientes con SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [36.] Mpl/Fragment 066 01 - Diskussion Bearbeitet: 28. November 2014, 18:05 Singulus Erstellt: 9. November 2014, 10:02 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 66, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 72, 73, Zeilen: 72: 16 ss. ; 73: 1 ss. |
|---|---|
| Aplasia pura de la serie roja. La aplasia pura de la serie roja se ha descrito de forma excepcional en el SS (151), tan sólo se han descrito 2 casos en la literatura (151,164) (Tabla 6), aunque existen otros casos adicionales en asociación con otras enfermedades autoinmunes como el LES y la AR. Los pacientes suelen presentar una anemia normocítica y normocrómica grave, asociada a reticulopenia y ausencia de eritroblastos en una médula ósea normal (164). La etiología de este síndrome hematológico en asociación con el SSp se desconoce, aunque parece ser que los mecanismos autoinmunes podrían ser los responsables del desarrollo de esta anemia, pues estudios in vitro han implicado al inhibidor eritropoyético IgG en suero y/o linfocitos citotóxicos como mediadores de la aplasia eritrocitaria (151,164). Sin embargo, algunos autores han puesto en duda una posible asociación (165). Esta inhibición afectaría a las células eritroides en diferentes etapas del desarrollo. Como los pacientes con SSp tienen un riesgo elevado de desarrollar linfomas, y considerando que la aplasia de la serie eritrocitaria puede ser la primera manifestación de una leucemia (linfocítica o linfoblástica) o de un linfoma (Hodgkin o no Hodgkin), es importante realizar un seguimiento clínico estricto de estos pacientes (164).
Mielodisplasia. Se han descrito pocos casos de pacientes con síndrome mielodisplásico en relación con el SS e incluso con el LES. Ramakrishna et al (151), describieron 2 pacientes ancianos con SS y anemia refractaria con sideroblastos en anillo. La presencia del síndrome mielodisplásico podría ser un hecho casual no relacionado con el SS pero sí con su edad avanzada. El síndrome mielodisplásico puede acompañarse de diversas anormalidades inmunológicas, incluyendo la presencia de ANA, aunque no hay estudios que relacionen este hecho con la asociación de enfermedades autoinmunes. |
Aplasia pura de la serie roja. La aplasia pura de la serie roja se ha descrito de forma excepcional en el SS (151), tan sólo se han descrito 2 casos en la literatura (151,164) (Tabla 6), aunque existen otros casos adicionales en asociación con otras enfermedades autoinmunes como el LES y la AR. Los pacientes suelen presentar una anemia normocítica y normocrómica grave, asociada a reticulopenia y ausencia de eritroblastos en una médula ósea normal (164). La etiología de este síndrome hematológico en asociación con el SSp se desconoce, aunque parece ser que los mecanismos autoinmunes podrían ser los responsables del desarrollo de esta anemia, pues estudios in vitro han implicado al inhibidor eritropoyético IgG en suero y/o linfocitos citotóxicos como mediadores de
[página 73] la aplasia eritrocitaria (151,164). Sin embargo, algunos autores han puesto en duda una posible asociación (165). Esta inhibición afectaría a las células eritroides en diferentes etapas del desarrollo. Como los pacientes con SSp tienen un riesgo elevado de desarrollar linfomas, y considerando que la aplasia de la serie eritrocitaria puede ser la primera manifestación de una leucemia (linfocítica o linfoblástica) o de un linfoma (Hodgkin o no Hodgkin), es importante realizar un seguimiento clínico estricto de estos pacientes (164). Mielodisplasia. Se han descrito pocos casos de pacientes con síndrome mielodisplásico en relación con el SS e incluso con el LES. Ramakrishna et al (151), describieron 2 pacientes ancianos con SS y anemia refractaria con sideroblastos en anillo. La presencia del síndrome mielodisplásico podría ser un hecho casual no relacionado con el SS pero sí con su edad avanzada. El síndrome mielodisplásico puede acompañarse de diversas anormalidades inmunológicas, incluyendo la presencia de ANA, aunque no hay estudios que relacionen este hecho con la asociación de enfermedades autoinmunes. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [37.] Mpl/Fragment 067 01 - Diskussion Bearbeitet: 28. November 2014, 18:15 Singulus Erstellt: 9. November 2014, 10:03 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 67, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 73, 74, Zeilen: 73: 16 ss. ; 74: 1 ss. |
|---|---|
| Anemia Perniciosa. La anemia perniciosa es infrecuente en el SS, aunque suele ser la consecuencia final de una gastritis atrófica crónica, que es la afección gastrointestinal más frecuente dentro del SSp (163). La asociación entre anemia perniciosa y SS fue descrita inicialmente por Wegelius et al (162) en 1950. En 1970, Williamson et al (161) describieron 2 pacientes con síndrome seco (xeroftalmía y xerostomía) y anemia perniciosa. Pedro Bodet et al (163) publicaron en 1993 otro caso adicional.
En la patogenia de la gastritis atrófica crónica en el SS, es probable que la inflamación crónica gástrica, conduzca a atrofia y consecuentemente a una anemia perniciosa. Sin embargo, la gastritis atrófica crónica no es suficiente para el desarrollo de una anemia perniciosa (163). La razón de esto se desconoce, aunque puede ser debido al patrón parcheado de la gastritis atrófica crónica en la mayoría de los casos, dejando así zonas libres de afección. Algunos estudios sugieren que la gastritis atrófica crónica en el SS está causada por la propia enfermedad de base ya que se han encontrado infiltrados mononucleares en la mucosa gástrica (163). Por otra parte, los anticuerpos anti-célula parietal (anti-CP) podrían tener un significado diagnóstico en la enfermedad, aunque su frecuencia en el SS varía ampliamente, entre el 10 y el 50%. b) Alteraciones de la serie blanca Entre un 12 y un 33% de los pacientes con SS pueden presentar leucopenia (12,129,130,132,166) (Tabla 8). |
Anemia Perniciosa. La anemia perniciosa es infrecuente en el SS, aunque suele ser la consecuencia final de una gastritis atrófica crónica, que es la afección gastrointestinal más frecuente dentro del SSp (163). La asociación entre anemia perniciosa y SS fue descrita inicialmente por Wegelius et al (162) en 1950. En 1970, Williamson et al (161) describieron 2 pacientes con síndrome seco (xeroftalmía y xerostomía) y anemia perniciosa. Pedro Bodet et al (163) publicaron en 1993 otro caso adicional.
En la patogenia de la gastritis atrófica crónica en el SS, es probable que la inflamación crónica gástrica, conduzca a atrofia y consecuentemente a una [página 74] anemia perniciosa. Sin embargo, la gastritis atrófica crónica no es suficiente para el desarrollo de una anemia perniciosa (163). La razón de esto se desconoce, aunque puede ser debido al patrón parcheado de la gastritis atrófica crónica en la mayoría de los casos, dejando así zonas libres de afección. Algunos estudios sugieren que la gastritis atrófica crónica en el SS está causada por la propia enfermedad de base ya que se han encontrado infiltrados mononucleares en la mucosa gástrica (163). Por otra parte, los anticuerpos anti-célula parietal (anti-CP) podrían tener un significado diagnóstico en la enfermedad, aunque su frecuencia en el SS varía ampliamente, entre el 10 y el 50%. b) Alteraciones de la serie blanca Entre un 12 y un 33% (125) de los pacientes con SS pueden presentar leucopenia (12,129,130,132,166) (Tabla 8). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [38.] Mpl/Fragment 068 01 - Diskussion Bearbeitet: 28. November 2014, 18:47 Singulus Erstellt: 9. November 2014, 10:08 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 68, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 74, 75, Zeilen: 74: 14 ss. ; 75: 1 ss. |
|---|---|
| TABLA 8. Prevalencia de leucopenia en pacientes con SS
Bloch et al (12) encontraron una prevalencia de leucopenia del 32% (20 de 62 pacientes), la mayoría con cifras cercanas a 3.000/mm3, aunque en 8 pacientes oscilaba entre 1500 - 2400/mm3. Alexander et al (130) encontraron una prevalencia del 23% de leucopenia (17 pacientes de 75), de los cuales, 14 mostraban anti-Ro/SSA positivo. En el estudio de Aoki et al (166), se encontró una prevalencia similar (26%), mientras que Skopouli et al (132) en un estudio de 261 pacientes, encontraron una prevalencia de leucopenia del 12%. Cabe destacar que la asociación de leucopenia y síndrome de Felty se ha descrito en algunos casos, especialmente en pacientes con SS asociado a AR (125). Linfocitos. Se ha descrito la existencia tanto de linfocitosis como de linfopenia en el SSp. La presencia de linfocitosis en el SS se presenta en un 10 y 40% (125). Bloch et al (12), encontraron linfocitosis en 7 pacientes de 62 (11.3%). Por otro lado, Aoki et al (166), encontraron linfopenia en 35 de 99 pacientes (35.3%). En este estudio, los pacientes con linfopenia mostraron menor frecuencia de artralgias aunque la presencia de anti-Ro/SSA y anti-La/SSB fue más frecuente, mientras que otros autores han reportado linfopenia absoluta en pacientes con SS principalmente asociado a AR. La causa de la linfopenia en el SS se desconoce. Henriksson et al (167), en un estudio de 214 pacientes con SSp, fueron los primeros en demostrar la presencia de anticuerpos [anti-CD4 en los lecucocitos de1 12% de pacientes con SSp.] |
TABLA 8. Prevalencia de leucopenia en pacientes con SS
Bloch et al (12) encontraron una prevalencia de leucopenia del 32% (20 de 62 pacientes), la mayoría con cifras cercanas a 3.000/mm3, aunque en 8 [página 75] pacientes oscilaba entre 1500 - 2400/mm3. Alexander et al (130) encontraron una prevalencia del 23% de leucopenia (17 pacientes de 75), de los cuales, 14 mostraban anti-Ro/SSA positivo. En el estudio de Aoki et al (166), se encontró una prevalencia similar (26%), mientras que Skopouli et al (132) en un estudio de 261 pacientes, encontraron una prevalencia de leucopenia del 12%. Cabe destacar que la asociación de leucopenia y síndrome de Felty se ha descrito en algunos casos, especialmente en pacientes con SS asociado a AR (125). Linfocitos. Se ha descrito la existencia tanto de linfocitosis como de linfopenia en el SSp. La presencia de linfocitosis en el SS se presenta en un 10 y 40% (125). Bloch et al (12), encontraron linfocitosis en 7 pacientes de 62 (11.3%). Por otro lado, Aoki et al (166), encontraron linfopenia en 35 de 99 pacientes (35.3%). En este estudio, los pacientes con linfopenia mostraron menor frecuencia de artralgias aunque la presencia de anti-Ro/SSA y anti-La/SSB fue más frecuente, mientras que otros autores han reportado linfopenia absoluta en pacientes con SS principalmente asociado a AR. La causa de la linfopenia en el SS se desconoce. Henriksson et al (167), en un estudio de 214 pacientes con SSp, fueron los primeros en demostrar la presencia de anticuerpos anti-CD4 en los lecucocitos de1 12% de pacientes con SSp. |
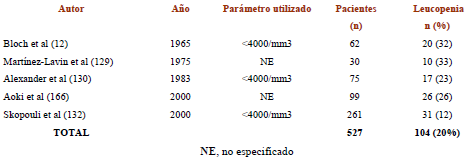
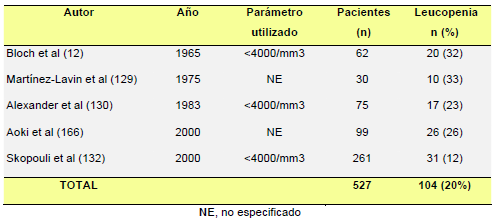
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [39.] Mpl/Fragment 069 01 - Diskussion Bearbeitet: 28. November 2014, 18:50 Singulus Erstellt: 9. November 2014, 10:10 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 69, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 75, 76, Zeilen: 75: 18 ss.; 76: 1 ss. |
|---|---|
| Sin embargo, no se encontró correlación alguna entre la presencia de anticuerpos anti-CD4 y la linfopenia. La presencia de anticuerpos anti-CD4 en pacientes con SS es una característica frecuente en pacientes con infección por el VIH y SS, lo que apoya una posible implicación vírica en la patogenia de la enfermedad.
Eosinófilos. La eosinofilia es una anomalía descrita en las series clásicas en aproximadamente un tercio de los pacientes con SS (30%) (125). Bloch et al (12), encontraron una prevalencia similar en un estudio de 62 pacientes: 27 presentaban más de 3% de eosinófilos; 16 más de 6% y 5 más de 10%, aunque sólo un paciente tuvo eosinofilias repetidas superiores al 30%. Es relevante destacar que ante la coexistencia de linfopenia grave con eosinofilia debe descartarse la existencia de un síndrome hipereosinofílico asociado (125). Neutrófilos. La neutropenia es una manifestación poco frecuente dentro del SS, siendo más frecuente en enfermedades como la AR y el LES. Se han publicado 6 casos de neutropenia en el SSp, 4 en hombres y 2 en mujeres (151,152,168-171) (Tabla 9). TABLA 9. Pacientes con SSp y neutropenia: casos publicados
|
Sin embargo, no se encontró correlación alguna entre la presencia de anticuerpos anti-CD4 y la linfopenia. La presencia de anticuerpos anti-CD4 en pacientes con SS es una característica frecuente en pacientes con infección por el VIH y SS, lo que apoya una posible implicación vírica en la patogenia de la enfermedad.
[página 76] Eosinófilos. La eosinofilia es una anomalía descrita en las series clásicas en aproximadamente un tercio de los pacientes con SS (30%) (125). Bloch et al (12), encontraron una prevalencia similar en un estudio de 62 pacientes: 27 presentaban más de 3% de eosinófilos; 16 más de 6% y 5 más de 10%, aunque sólo un paciente tuvo eosinofilias repetidas superiores al 30%. Es relevante destacar que ante la coexistencia de linfopenia grave con eosinofilia debe descartarse la existencia de un síndrome hipereosinofílico asociado (125). Neutrófilos. La neutropenia es una manifestación poco frecuente dentro del SS, siendo más frecuente en enfermedades como la AR y el LES. Se han publicado 6 casos de neutropenia en el SSp, 4 en hombres y 2 en mujeres (151,152,168-171) (Tabla 9). TABLA 9. Pacientes con SSp y neutropenia: casos publicados
|
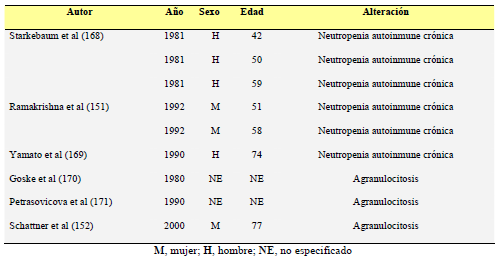
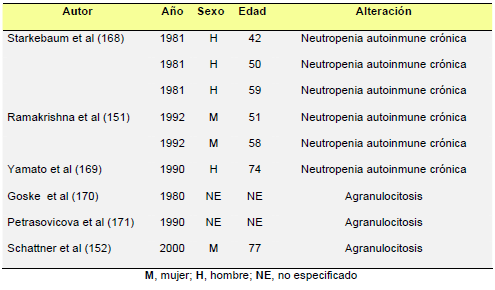
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [40.] Mpl/Fragment 070 01 - Diskussion Bearbeitet: 28. November 2014, 18:53 Singulus Erstellt: 9. November 2014, 10:12 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 70, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 77, Zeilen: 1 ss. |
|---|---|
| Aunque estudios previos ya habían descrito de forma aislada la existencia de anticuerpos antineutrófilo en pacientes con SSp (172), fue en 1995 (173) cuando se analizó de manera específica su presencia en una serie de 66 pacientes, encontrándose una prevalencia del 45%. Los autores no encontraron correlación entre la presencia de anticuerpos con el recuento de neutrófilos, sugiriendo que los autoanticuerpos se producen como consecuencia de la liberación del FcγRIIIb que sigue a la activación de los polimorfonucleares (PMN). Aunque la neutropenia crónica es un hallazgo infrecuente en el SSp (151), de manera puntual se ha correlacionado con la existencia de anticuerpos antineutrófilo (169). En otro estudio, Lamour et al (174) analizaron la presencia de anticuerpos IgG e IgM contra el FcγRIII en 66 pacientes con SSp, encontrando una prevalencia del 24% y 15%, respectivamente. Estos autores encontraron una correlación significativa entre la neutropenia y la presencia de artritis no erosiva, fenómeno de Raynaud, afección pulmonar y la presencia del alelo HLA-DR3. Sin embargo, no encontraron correlación con el nivel sérico de IgG ni con el recuento sérico de PMN. Finalmente, se han detectado niveles circulantes de FcγRIII en pacientes con SSp (175), ya que el FcγRIII puede liberarse tras la activación de los PMN. En conclusión, no es posible establecer con claridad el papel de los anticuerpos antipolimorfonucleares en el SSp con los datos que se disponen en la actualidad.
Finalmente, la granulocitopenia inmune grave o agranulocitosis es poco habitual en las enfermedades autoinmunes en general y en particular en el SSp donde sólo se han reportado 3 casos en la literatura (152,170,171) (Tabla 9). |
Aunque estudios previos ya habían descrito de forma aislada la existencia de anticuerpos antineutrófilo en pacientes con SSp (172), fue en 1995 (173) cuando se analizó de manera específica su presencia en una serie de 66 pacientes, encontrándose una prevalencia del 45%. Los autores no encontraron correlación entre la presencia de anticuerpos con el recuento de neutrófilos, sugiriendo que los autoanticuerpos se producen como consecuencia de la liberación del FcγRIIIb que sigue a la activación de los polimorfonucleares (PMN). Aunque la neutropenia crónica es un hallazgo infrecuente en el SSp (151), de manera puntual se ha correlacionado con la existencia de anticuerpos antineutrófilo (169). En otro estudio, Lamour et al (174) analizaron la presencia de anticuerpos IgG e IgM contra el FcγRIII en 66 pacientes con SSp, encontrando una prevalencia del 24% y 15%, respectivamente. Estos autores encontraron una correlación significativa entre la neutropenia y la presencia de artritis no erosiva, fenómeno de Raynaud, afección pulmonar y la presencia del alelo HLA-DR3. Sin embargo, no encontraron correlación con el nivel sérico de IgG ni con el recuento sérico de PMN. Finalmente, se han detectado niveles circulantes de FcγRIII en pacientes con SSp (175), ya que el FcγRIII puede liberarse tras la activación de los PMN. En conclusión, no es posible establecer con claridad el papel de los anticuerpos antipolimorfonucleares en el SSp con los datos que se disponen en la actualidad.
Finalmente, la granulocitopenia inmune grave o agranulocitosis es poco habitual en las enfermedades autoinmunes en general y en particular en el SSp donde sólo se han reportado 3 casos en la literatura (152,170,171) (Tabla 9). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [41.] Mpl/Fragment 071 01 - Diskussion Bearbeitet: 6. December 2014, 10:43 Singulus Erstellt: 9. November 2014, 10:17 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 71, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 78, 79, Zeilen: 78: 1 ss. ; 79: 1 ss. |
|---|---|
| c) Alteraciones de la serie plaquetaria
La alteración plaquetaria en el SSp se manifiesta generalmente como una trombocitopenia leve (100 – 150,000/mm3) con una prevalencia que oscila entre el 8 y el 15% según diversos estudios (12,130,151,166) (Tabla 10) Bloch et al (12) encontraron trombocitopenia leve (100 – 150,000/mm3) en 5 de 62 pacientes (8%) mas ningún caso de trombocitopenia severa (<50,000/mm3). Ramakrishna et al (151) en un estudio de 27 pacientes con SS describieron 5 con trombocitopenia (19%), 4 de ellos diagnosticados de púrpura trombocitopénica idiopática y dos de ellos con una buena respuesta al tratamiento corticosteroide. Una revisión de la literatura japonesa (176), ha reportado más casos de trombocitopenia en pacientes con SSp que en el SS asociado. De los 20 pacientes con SS estudiados, la trombocitopenia se presentó en 16 con SSp y en 4 con SS asociado. TABLA 10. Prevalencia de trombocitopenia en pacientes con SS
Por otra parte, se considera que la trombocitopenia grave (<50,000/mm3) es infrecuente dentro del SSp, tan sólo se han descrito 6 casos de trombocitopenia grave (151,152,176) (Tabla 11). Entre ellos destacan 5 mujeres (entre los 37 y los 66 años) y un varón de 9 años, en quienes se constatan datos de sangrado espontáneo; los cinco pacientes fueron tratados con corticoides, además de ciclofosfamida y plasmaféresis en la única paciente [que presentó diátesis hemorrágica.] |
c) Alteraciones de la serie plaquetaria
La alteración plaquetaria en el SSp se manifiesta generalmente como una trombocitopenia leve (100 – 150 000/mm3) con una prevalencia que oscila entre el 8 y el 15% según diversos estudios (12,130,151,166) (Tabla 10) TABLA 10. Prevalencia de trombocitopenia en pacientes con SS
Bloch et al (12) encontraron trombocitopenia leve (100 – 150,000/mm3) en 5 de 62 pacientes (8%) mas ningún caso de trombocitopenia severa (<50,000/mm3). Ramakrishna et al (151) en un estudio de 27 pacientes con SS describieron 5 con trombocitopenia (19%), 4 de ellos diagnosticados de púrpura trombocitopénica idiopática y dos de ellos con una buena respuesta al tratamiento corticosteroide. Una revisión de la literatura japonesa (176), ha reportado más casos de trombocitopenia en pacientes con SSp que en el SS asociado. De los 20 pacientes con SS estudiados, la trombocitopenia se presentó en 16 con SSp y en 4 con SS asociado. [página 79] Por otra parte, se considera que la trombocitopenia grave (<50,000/mm3) es infrecuente dentro del SSp, tan sólo se han descrito 6 casos de trombocitopenia grave (151,152,176) (Tabla 11). [...] Entre ellos destacan 5 mujeres (entre los 37 y los 66 años) y un varón de 9 años, en quienes se constatan datos de sangrado espontáneo; los cinco pacientes fueron tratados con corticoides, además de ciclofosfamida y plasmaféresis en la única paciente que presentó diátesis hemorrágica. |
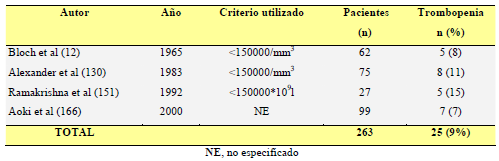
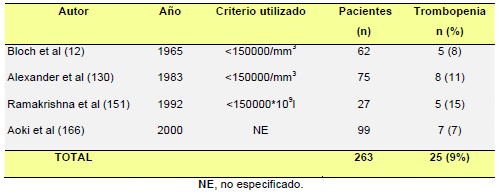
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [42.] Mpl/Fragment 072 01 - Diskussion Bearbeitet: 6. December 2014, 10:46 Singulus Erstellt: 9. November 2014, 10:19 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 72, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 79, 80, Zeilen: 79: 7 ss. ; 80: 1 ss. |
|---|---|
| Por otra parte, se ha descrito un caso de trombocitopenia severa de causa farmacológica en un paciente con SS. Haro et al (177), describieron un paciente con SS y fallo cardiaco, el cual desarrolló trombocitopenia grave tras la aplicación de digoxina. Esta revirtió tras la suspención inmediata del medicamento. La coexistencia de un SSp con otros procesos plaquetopénicos como la púrpura trombótica trombocitopénica debe considerarse como un hecho excepcional. La trombocitopenia en pacientes con SS está principalmente causada por la destrucción plaquetaria debida a anticuerpos antiplaquetarios o por la mediación de inmunocomplejos, similar al de los pacientes con LES. En 3 pacientes con SS y plaquetopenia descritos en la literatura, se han detectado anticuerpos antiplaquetarios IgG y anticuerpos IgM en 1 caso (176). La trombocitopenia se ha relacionado con la presencia de anticuerpos anti-Ro/SSA y/o anticuerpos anti-La/SSB. Alexander et al (130) encontraron trombocitopenia en 11% de los pacientes (8 de 75) con SS, siete de ellos con anti-Ro/SSA y Aoki et al (166) encontraron trombocitopenia en 7 de sus 99 (7%) pacientes, todos con anticuerpos anti-La/SSB. Es de importancia mencionar que esta anormalidad hematológica fue más frecuente en pacientes jóvenes y varones que en mujeres, los cuales mostraron mayor tendencia a la afección cutánea, a la presencia de ANA y FR. | Por otra parte, se ha descrito un caso de trombocitopenia severa de causa farmacológica en un paciente con SS. Haro et al (177), describieron un paciente con SS y fallo cardiaco, el cual desarrolló trombocitopenia grave tras la aplicación de digoxina. Esta revirtió tras la suspención inmediata del medicamento. La coexistencia de un SSp con otros procesos plaquetopénicos
[página 80] como la púrpura trombótica trombocitopénica debe considerarse como un hecho excepcional. La trombocitopenia en pacientes con SS está principalmente causada por la destrucción plaquetaria debida a anticuerpos antiplaquetarios o por la mediación de inmunocomplejos, similar al de los pacientes con LES. En 3 pacientes con SS y plaquetopenia descritos en la literatura, se han detectado anticuerpos antiplaquetarios IgG y anticuerpos IgM en 1 caso (176). La trombocitopenia se ha relacionado con la presencia de anticuerpos anti-Ro/SSA y/o anticuerpos anti-La/SSB. Alexander et al (130) encontraron trombocitopenia en 11% de los pacientes (8 de 75) con SS, siete de ellos con anti-Ro/SSA y Aoki et al (166) encontraron trombocitopenia en 7 de sus 99 (7%) pacientes, todos con anticuerpos anti-La/SSB. Es de importancia mencionar que esta anormalidad hematológica fue más frecuente en pacientes jóvenes y varones que en mujeres, los cuales mostraron mayor tendencia a la afección cutánea, a la presencia de ANA y FR. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [43.] Mpl/Fragment 073 01 - Diskussion Bearbeitet: 6. December 2014, 10:50 Singulus Erstellt: 9. November 2014, 10:22 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 73, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 79, 80, 81, Zeilen: 79: tabla; 80: 15 ss.; 81: 1 ss. |
|---|---|
| TABLA 11. Trombocitopenia grave (<50,000) en pacientes con SSp
En conclusión, la trombocitopenia, al igual que cualquier otra anormalidad hematológica, puede constituir el primer signo de un SS latente (152,178,179) bien sea primario o asociado. 1.5. Alteraciones Inmunológicas 1.5.1. Anticuerpos antinucleares La determinación de los ANA suele ser el principal dato inmunológico a solicitar cuando se sospecha la existencia de una enfermedad autoinmune. Su positividad forma parte de los criterios diagnósticos tanto del LES como del SSp. La positividad de los ANA en pacientes con SSp suele ser superior al 80% (180), aunque los trabajos publicados respecto a su asociación con manifestaciones clínicas son escasos. Nuestro grupo (180) no ha encontrado relación estadísticamente significativa con ninguna manifestación clínica y sí con la presencia de anticuerpos anti-Ro/SSA y anti-La/SSB. |
TABLA 11. Trombocitopenia grave (<50,000) en pacientes con SSp
[página 80] En conclusión, la trombocitopenia, al igual que cualquier otra anormalidad hematológica, puede constituir el primer signo de un SS latente (152,178,179) bien sea primario o asociado. 1.5. Alteraciones Inmunológicas 1.5.1. Anticuerpos antinucleares La determinación de los ANA suele ser el principal dato inmunológico a solicitar cuando se sospecha la existencia de una enfermedad autoinmune. Su positividad forma parte de los criterios diagnósticos tanto del LES como del SSp. La positividad de los ANA en pacientes con SSp suele ser superior al 80% (180), aunque los trabajos publicados respecto a su asociación con manifestaciones [página 80] clínicas son escasos. Nuestro grupo (180) no ha encontrado relación estadísticamente significativa con ninguna manifestación clínica y sí con la presencia de anticuerpos anti-Ro/SSA y anti-La/SSB. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [44.] Mpl/Fragment 074 01 - Diskussion Bearbeitet: 6. December 2014, 17:36 Singulus Erstellt: 9. November 2014, 10:24 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 74, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 81, 82, Zeilen: 81: 3 ss.; 82: 1 ss. |
|---|---|
| [En pacientes con títulos] elevados de ANA y negatividad para anti-Ro/La, hemos detectado (97) su asociación con mayor frecuencia de afección pulmonar y fenómeno de Raynaud.
1.5.2. FR El FR es una inmunoglobulina IgM dirigida contra la fracción Fc de inmunoglobulinas IgG autólogas circulantes. En la mayoría de los estudios realizados en pacientes con SSp se ha demostrado un porcentaje elevado de positividad para el FR (181), cercana al 50%. Sin embargo, son pocos los trabajos que han analizado la asociación entre la presencia de FR y las manifestaciones clínicas e inmunológicas del SSp. En un estudio (95), se ha encontrado una prevalencia del 38%, destacando una relación estadísticamente significativa con la presencia de afección articular, vasculitis cutánea y positividad para anticuerpos anti-Ro/SSA y anti-La/SSB. Es posible que en algunos de los pacientes con SSp la detección de FR se asocie a la presencia de crioglobulinemia, reflejando la actividad de tipo FR que poseen dichas crioglobulinas. 1.5.3. Anticuerpos anti-Ro/SSA y anti-La/SSB Los anticuerpos contra el antígeno Ro/SSA se describieron por primera vez en 1962 en el suero de pacientes con SSp. Se consideran los autoanticuerpos con mayor especificidad para el diagnóstico del SSp, aunque aparezcan en porcentajes variables (30 - 70%, según la técnica empleada y la cohorte estudiada). Por otra parte, existe una clara relación entre la existencia de anticuerpos anti-Ro/SSA maternos y bloqueo cardíaco congénito (182). El primer dato sugestivo de SSp en mujeres Ro positivas asintomáticas puede ser el nacimiento de un niño con bloqueo cardíaco congénito, ya que el 60% de las madres están asintomáticas en el momento del nacimiento. También se ha descrito la asociación de miositis y positividad para anticuerpos anti-Ro/SSA en pacientes con SSp. |
En pacientes con títulos elevados de ANA y negatividad para anti-Ro/La, hemos detectado (97) su asociación con mayor frecuencia de afección pulmonar y fenómeno de Raynaud.
1.5.2. FR El FR es una inmunoglobulina IgM dirigida contra la fracción Fc de inmunoglobulinas IgG autólogas circulantes. En la mayoría de los estudios realizados en pacientes con SSp se ha demostrado un porcentaje elevado de positividad para el FR (181), cercana al 50%. Sin embargo, son pocos los trabajos que han analizado la asociación entre la presencia de FR y las manifestaciones clínicas e inmunológicas del SSp. En un estudio (95), se ha encontrado una prevalencia del 38%, destacando una relación estadísticamente significativa con la presencia de afección articular, vasculitis cutánea y positividad para anticuerpos anti-Ro/SSA y anti-La/SSB. Es posible que en algunos de los pacientes con SSp la detección de FR se asocie a la presencia de crioglobulinemia, reflejando la actividad de tipo FR que poseen dichas crioglobulinas. 1.5.3. Anticuerpos anti-Ro/SSA y anti-La/SSB Los anticuerpos contra el antígeno Ro/SSA se describieron por primera vez en 1962 en el suero de pacientes con SSp. Se consideran los autoanticuerpos con mayor especificidad para el diagnóstico del SSp, aunque aparezcan en porcentajes variables (30 - 70%, según la técnica empleada y la [página 82] cohorte estudiada). Por otra parte, existe una clara relación entre la existencia de anticuerpos anti-Ro/SSA maternos y bloqueo cardíaco congénito (182). El primer dato sugestivo de SSp en mujeres Ro positivas asintomáticas puede ser el nacimiento de un niño con bloqueo cardíaco congénito, ya que el 60% de las madres están asintomáticas en el momento del nacimiento. También se ha descrito la asociación de miositis y positividad para anticuerpos anti-Ro/SSA en pacientes con SSp. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [45.] Mpl/Fragment 075 01 - Diskussion Bearbeitet: 6. December 2014, 17:39 Singulus Erstellt: 9. November 2014, 10:26 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 75, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 82, 83, Zeilen: 82: 7 ss. ; 83: 1 ss. |
|---|---|
| Finalmente, se han descrito otras asociaciones. Alexander et al (183) han descrito una mayor prevalencia de parotidomegalia y linfadenopatía en pacientes con SSp y anticuerpos anti-Ro/SSA. También se ha descrito una mayor prevalencia de anti-Ro/SSA en aquellos pacientes con un inicio precoz de la enfermedad (184). Clásicamente se ha asociado la presencia de anticuerpos anti-Ro/SSA con la existencia de anemia (183,185), leucopenia (126,183,185-187), linfopenia (166,186,187) y trombocitopenia (166,185). Recientemente se ha corroborado la asociación con leucopenia en un análisis estadístico multivariado (188). Por otra parte, diversos autores han asociado la existencia de hipergammaglobulinemia con anticuerpos anti-Ro/SSA positivos (183,185).
Por otra parte, diversos estudios han analizado la relación entre los anticuerpos anti-La/SSB y las manifestaciones clínicas del SSp. Venables et al (189) encontraron una asociación entre el anti-La/SSB, parotidomegalia y el desarrollo de púrpura cutánea. Otros autores han descrito su asociación con la leucopenia, la linfopenia, la hipergammaglobulinemia, el FR, la parotidomegalia y la vasculitis (185). Por otra parte, se ha descrito mayor afección articular, fenómeno de Rauynaud, vasculitis cutánea, afección tiroidea y positividad para ANA, FR y anti-Ro/SSA en aquellos con anti-La/SSB, además de una posible asociación con linfopenia y trombocitopenia (188). 1.5.4. Otros autoanticuerpos Los anticuerpos contra el ácido desoxirribonucleico de doble cadena (anti-dsDNA) y los anti-Sm constituyen uno de los criterios diagnósticos de LES, y no suelen detectarse en pacientes con SSp. Estudios recientes sugieren que la existencia de títulos elevados de anti-dsDNA en pacientes con SSp sugiere una posible evolución a LES (190,191). Respecto a los anticuerpos contra la ribonucleoproteína (anti-RNP), se han descrito prevalencias que oscilan entre el 8 y el 28% de casos, aunque probablemente se incluyan [tanto SSp primarios como asociados.] |
Finalmente, se han descrito otras asociaciones. Alexander et al (183) han descrito una mayor prevalencia de parotidomegalia y linfadenopatía en pacientes con SSp y anticuerpos anti-Ro/SSA. También se ha descrito una mayor prevalencia de anti-Ro/SSA en aquellos pacientes con un inicio precoz de la enfermedad (184). Clásicamente se ha asociado la presencia de anticuerpos anti-Ro/SSA con la existencia de anemia (183,185), leucopenia (126,183,185- 187), linfopenia (166,186,187) y trombocitopenia (166,185). Recientemente se ha corroborado la asociación con leucopenia en un análisis estadístico multivariado (188). Por otra parte, diversos autores han asociado la existencia de hipergammaglobulinemia con anticuerpos anti-Ro/SSA positivos (183,185).
Por otra parte, diversos estudios han analizado la relación entre los anticuerpos anti-La/SSB y las manifestaciones clínicas del SSp. Venables et al (189) encontraron una asociación entre el anti-La/SS-B, parotidomegalia y el desarrollo de púrpura cutánea. Otros autores han descrito su asociación con la leucopenia, la linfopenia, la hipergammaglobulinemia, el FR, la parotidomegalia y la vasculitis (185). Por otra parte, se ha descrito mayor afección articular, fenómeno de Rauynaud, vasculitis cutánea, afección tiroidea y positividad para ANA, FR y anti-Ro/SS-A en aquellos con anti-La/SSB, además de una posible asociación con linfopenia y trombocitopenia (188). [página 83] 1.5.4. Otros autoanticuerpos Los anticuerpos contra el ácido desoxirribonucleico de doble cadena (anti-dsDNA) y los anti-Sm constituyen uno de los criterios diagnósticos de LES, y no suelen detectarse en pacientes con SSp. Estudios recientes sugieren que la existencia de títulos elevados de anti-dsDNA en pacientes con SSp sugiere una posible evolución a LES (190,191). Respecto a los anticuerpos contra la ribonucleoproteína (anti-RNP), se han descrito prevalencias que oscilan entre el 8 y el 28% de casos, aunque probablemente se incluyan tanto SSp primarios como asociados. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [46.] Mpl/Fragment 076 01 - Diskussion Bearbeitet: 6. December 2014, 17:42 Singulus Erstellt: 9. November 2014, 10:36 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 76, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 83, 84, Zeilen: 83: 9: ss. ; 84: 1 ss. |
|---|---|
| De forma aislada se ha descrito la positividad de anticuerpos anticentrómero (ACA) en pacientes con SSp. Es posible que su detección pueda representar un dato inicial de posible evolución a ES limitada. Es aconsejable detectar los ACA en todo paciente con SS y Raynaud, especialmente en aquellos con ANA a títulos elevados con negatividad para Ro/La. Varios autores han analizado la prevalencia de anticuerpos antifosfolipídicos (AAF) en pacientes con SSp, encontrando una prevalencia que oscila entre el 2 y el 33%, aunque su presencia suele considerarse como un mero epifenómeno autoinmune sin repercusiones clínicas. La presencia de anticuerpos AMA fue descrita por primera vez en 1965 en pacientes con CBP, en los que se detectan en un 95% de los casos. El frecuente solapamiento entre la CBP y el SSp motiva que la detección de AMA en un paciente con SSp sugiera la existencia de una CBP asociada (192).
1.5.5. Crioglobulinas Las crioglobulinas son inmunoglobulinas circulantes que precipitan in vitro con el frío. Un estudio reciente (144) ha analizado la presencia de crioglobulinas en 115 pacientes con SSp, encontrando una prevalencia del 16%, muy similar a la obtenida por Tzioufas et al (193) (19% en 103 pacientes). El papel de las crioglobulinas en el SSp se centra en tres importantes aspectos: su clara relación con manifestaciones clínicas de tipo vasculítico, su asociación con el VHC (obliga a descartar dicha infección en todo paciente con SSp y crioglobulinemia) y finalmente, su carácter predictivo de posible desarrollo a linfoma. 1.5.6. Hipocomplementemia La determinación rutinaria del complemento (C3, C4 y CH50) es una herramienta clínica importante en el manejo de algunas enfermedades autoinmunes [sistémicas.] |
De forma aislada se ha descrito la positividad de anticuerpos anticentrómero (ACA) en pacientes con SSp. Es posible que su detección pueda representar un dato inicial de posible evolución a ES limitada. Es aconsejable detectar los ACA en todo paciente con SS y Raynaud, especialmente en aquellos con ANA a títulos elevados con negatividad para Ro/La. Varios autores han analizado la prevalencia de anticuerpos antifosfolipídicos (AAF) en pacientes con SSp, encontrando una prevalencia que oscila entre el 2 y el 33%, aunque su presencia suele considerarse como un mero epifenómeno autoinmune sin repercusiones clínicas. La presencia de anticuerpos AMA fue descrita por primera vez en 1965 en pacientes con CBP, en los que se detectan en un 95% de los casos. El frecuente solapamiento entre la CBP y el SSp motiva que la detección de AMA en un paciente con SSp sugiera la existencia de una CBP asociada (192).
1.5.5. Crioglobulinas Las crioglobulinas son inmunoglobulinas circulantes que precipitan in vitro con el frío (Figura 7). Un estudio reciente (144) ha analizado la presencia de [página 84] crioglobulinas en 115 pacientes con SSp, encontrando una prevalencia del 16%, muy similar a la obtenida por Tzioufas et al (193) (19% en 103 pacientes). El papel de las crioglobulinas en el SSp se centra en tres importantes aspectos: su clara relación con manifestaciones clínicas de tipo vasculítico, su asociación con el VHC (obliga a descartar dicha infección en todo paciente con SSp y crioglobulinemia) y finalmente, su carácter predictivo de posible desarrollo a linfoma. [...] 1.5.6. Hipocomplementemia La determinación rutinaria del complemento (C3, C4 y CH50) es una herramienta clínica importante en el manejo de algunas enfermedades autoinmunes sistémicas. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [47.] Mpl/Fragment 077 01 - Diskussion Bearbeitet: 6. December 2014, 17:46 Singulus Erstellt: 9. November 2014, 13:04 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 77, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 84, 85, Zeilen: 84: 11 ss.; 85: 1 ss. |
|---|---|
| El mejor ejemplo es el LES, en el cual, la hipocomplementemia se correlaciona directamente con la actividad de la enfermedad, especialmente con el desarrollo de neuropatía. A pesar de que la hipocomplementemia es un dato inmunológico que con frecuencia presentan los pacientes con SSp, su estudio en esta enfermedad ha sido escaso. Suele observarse un descenso del CH50, sobretodo a expensas del descenso de C4, siendo más raro el descenso de C3. Suele traducir la existencia de una crioglobulinemia asociada, y se ha demostrado que está relacionada con una mayor frecuencia de procesos linfoproliferativos y mayor mortalidad en pacientes con SSp (132).
1.6. Diagnóstico 1.6.1. Estudio de la función salivar Por un lado, puede analizarse la cantidad de flujo salival y su composición. El estudio del flujo salival basal y estimulado con pilocarpina refleja el estado funcional parotídeo, y se correlaciona con los resultados de la gammagrafía parotídea y la biopsia salival. Para el estudio de la estructura anatómica se pueden utilizar técnicas ecográficas, sialográficas o gammagráficas. La sialografía suele provocar incomodidad y se substituye por la gammagrafía, que utiliza Tecnecio 99 y valora la captación y excreción del trazador, con unos criterios diagnósticos (Tabla 12) (Figura 3). La ecografía de las glándulas parótidas y submandibulares muestra áreas hipoecoicas y diversos grados de desestructuración que podrían corresponder a focos de infiltración linfocitaria (194). Recientemente se ha propuesto el estudio parotídeo mediante RM (195). |
El mejor ejemplo es el LES, en el cual, la hipocomplementemia se correlaciona directamente con la actividad de la enfermedad, especialmente con el desarrollo de neuropatía. A pesar de que la hipocomplementemia es un dato inmunológico que con frecuencia presentan los pacientes con SSp, su estudio en esta enfermedad ha sido escaso. Suele
[página 85] observarse un descenso del CH50, sobretodo a expensas del descenso de C4, siendo más raro el descenso de C3. Suele traducir la existencia de una crioglobulinemia asociada, y se ha demostrado que está relacionada con una mayor frecuencia de procesos linfoproliferativos y mayor mortalidad en pacientes con SSp (132). 1.6. Diagnóstico 1.6.1. Estudio de la función salivar Por un lado, puede analizarse la cantidad de flujo salival y su composición. El estudio del flujo salival basal y estimulado con pilocarpina refleja el estado funcional parotídeo, y se correlaciona con los resultados de la gammagrafía parotídea y la biopsia salival. Para el estudio de la estructura anatómica se pueden utilizar técnicas ecográficas, sialográficas o gammagráficas. La sialografía suele provocar incomodidad y se substituye por la gammagrafía, que utiliza Tecnecio 99 y valora la captación y excreción del trazador, con unos criterios diagnósticos (Tabla 12) (Figura 8). La ecografía de las glándulas parótidas y submandibulares muestra áreas hipoecoicas y diversos grados de desestructuración que podrían corresponder a focos de infiltración linfocitaria (194) Recientemente se ha propuesto el estudio parotídeo mediante RM (195) (Figura 9). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [48.] Mpl/Fragment 078 01 - Diskussion Bearbeitet: 6. December 2014, 17:49 Singulus Erstellt: 9. November 2014, 13:07 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 78, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 80, Zeilen: 86: 1 ss. ; 87: 1 ss. |
|---|---|
| TABLA 12. Criterios gammagráficos de afección de las glándulas salivales
FIGURA 3. Gammagrafía salival en un paciente con síndrome de Sjögren. Ausencia de captación del radiotrazador en las glándulas submaxilares y disminución de la concentración del mismo en las parótidas
1.6.2. Estudio de la función lagrimal La atrofia glandular lagrimal origina disminución de la secreción lagrimal (hipolagrimación) y una lesión descamativa del epitelio conjuntival y corneal. Las pruebas diagnósticas estudian por un lado la secreción lagrimal y por otro el estado del epitelio corneal. |
TABLA 12. Criterios gammagráficos de afección de las glándulas salivales
FIGURA 8. Gammagrafía salival en un paciente con síndrome de Sjögren. Ausencia de captación del radiotrazador en las glándulas submaxilares y disminución de la concentración del mismo en las parótidas
[página 87] [...] 1.6.2. Estudio de la función lagrimal La atrofia glandular lagrimal origina disminución de la secreción lagrimal (hipolagrimación) y una lesión descamativa del epitelio conjuntival y corneal. Las pruebas diagnósticas estudian por un lado la secreción lagrimal y por otro el estado del epitelio corneal. |

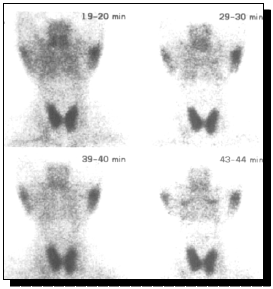
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [49.] Mpl/Fragment 079 01 - Diskussion Bearbeitet: 6. December 2014, 17:56 Singulus Erstellt: 9. November 2014, 13:10 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 79, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 87, 88, 89, Zeilen: 87: 5 ss.; 88: 1 ss. ; 89: 1 ss. |
|---|---|
| [Para la prueba de Schirmer se utiliza un papel de filtro de 35 mm de largo por 5] mm de ancho que se adapta al canto externo del párpado inferior. Se lee a los 5 minutos considerándose una prueba cuantitativa de hiposecreción basal cuando es inferior a los 5 mm. La tinción con rosa de Bengala es una prueba cualitativa que valora las alteraciones de la capa mucínica después de aplicar en la córnea un colorante (rosa de Bengala al 1%) en el fórnix conjuntival inferior.
1.6.3. Histopatología La biopsia salival permite valorar la estructura glandular y la infiltración inflamatoria (Figura 4). El estudio de otras glándulas exocrinas ha revelado la similitud con los hallazgos de la biopsia salival. El infiltrado linfocitario está constituido por linfocitos T CD4 (45-55%), linfocitos T supresores/citotóxicos CD8 (10-20%) y linfocitos B (20-35%). Los infiltrados celulares se localizan principalmente en los ductus y aparentemente se extienden a los acinos. La pérdida de los acinos es la anormalidad parenquimatosa dominante y se asocia de manera significativa al tamaño de los focos infiltrativos. Los indicadores de actividad linfocitaria son: mayor tamaño o número de infiltrados, aparición de centros germinales e infiltración de las vénulas postcapilares del endotelio. En las fases avanzadas hay atrofia y sustitución adiposa del parénquima glandular. La interpretación de la biopsia salival se realiza siguiendo los criterios propuestos por Chisholm y Mason (13). La gradación histológica del número y tamaño de los infiltrados linfocitarios de las glándulas salivales “focus score” es el principal marcador de la afección exocrina del SS. |
Para la prueba de Schirmer se utiliza un papel de filtro de 35 mm de largo por 5 mm de ancho que se adapta al canto externo del párpado inferior. Se lee a los 5 minutos considerándose una prueba cuantitativa de hiposecreción basal cuando es inferior a los 5 mm. La tinción con rosa de Bengala es una prueba cualitativa que valora las alteraciones de la capa mucínica después de aplicar en la córnea un colorante (rosa de Bengala al 1%) en el fórnix conjuntival inferior (Figura 10).
[página 88] [...] 1.6.3. Histopatología La biopsia salival permite valorar la estructura glandular y la infiltración inflamatoria (Figura 11). El estudio de otras glándulas exocrinas ha revelado la similitud con los hallazgos de la biopsia salival. El infiltrado linfocitario está constituido por linfocitos T CD4 (45-55%), linfocitos T supresores/citotóxicos CD8 (10-20%) y linfocitos B (20-35%). Los infiltrados celulares se localizan principalmente en los ductus y aparentemente se extienden a los acinos. La pérdida de los acinos es la anormalidad parenquimatosa dominante y se asocia de manera significativa al tamaño de los focos infiltrativos. Los indicadores de actividad linfocitaria son: mayor tamaño o número de infiltrados, aparición de centros germinales e infiltración de las vénulas postcapilares del endotelio. En las fases avanzadas hay atrofia y sustitución adiposa del parénquima glandular. La interpretación de la biopsia salival se realiza siguiendo los criterios propuestos por Chisholm y Mason (13). La gradación histológica del número y tamaño de los [página 89] infiltrados linfocitarios de las glándulas salivales “focus score” es el principal marcador de la afección exocrina del SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [50.] Mpl/Fragment 080 01 - Diskussion Bearbeitet: 6. December 2014, 18:00 Singulus Erstellt: 9. November 2014, 13:48 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 80, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 89, 90, Zeilen: 89: 3 ss.; 90: 1 ss. |
|---|---|
| FIGURA 4. Infiltrado linfocitario en una biopsia salival
1.6.4. Criterios clasificatorios Existe cierta controversia respecto a los criterios clasificatorios del SS, especialmente entre autores americanos y europeos, por lo que se han propuesto varias clasificaciones (196). En Europa suelen utilizarse los criterios propuestos en 1993 por el Grupo de Estudio de la Comunidad Europea para el SS, aunque recientemente se ha producido un consenso Europeo-Americano que modifica dichos criterios. Los Criterios del Grupo de Estudio de la Comunidad Europea (CE) (197) (Tabla 13) son unos criterios realizados en base a un amplio número de pacientes y centros de diversos países europeos. Para el criterio histológico se utiliza la presencia de un foco como dato patológico, mientras en los de San Diego se requieren dos o más focos (198). La presencia de autoanticuerpos está incluida en los CE, pero sólo el 36% de los pacientes incluidos tenían anti-Ro positivos, mientras que con los criterios de San Diego lo tenían el 90% de los pacientes. |
FIGURA 11. Infiltrado linfocitario en una biopsia salival
1.6.4. Criterios clasificatorios Existe cierta controversia respecto a los criterios clasificatorios del SS, especialmente entre autores americanos y europeos, por lo que se han propuesto varias clasificaciones (196). En Europa suelen utilizarse los criterios propuestos en 1993 por el Grupo de Estudio de la Comunidad Europea para el SS, aunque recientemente se ha producido un consenso Europeo-Americano que modifica dichos criterios. Los Criterios del Grupo de Estudio de la Comunidad Europea (CE) (197) (Tabla 13) son unos criterios realizados en base a un amplio número de pacientes y centros de diversos países europeos. Para el criterio histológico se utiliza la presencia de un foco como dato patológico, mientras en los de San Diego se [página 90] requieren dos o más focos (198). La presencia de autoanticuerpos está incluida en los CE, pero sólo el 36% de los pacientes incluidos tenían anti-Ro positivos, mientras que con los criterios de San Diego lo tenían el 90% de los pacientes. |
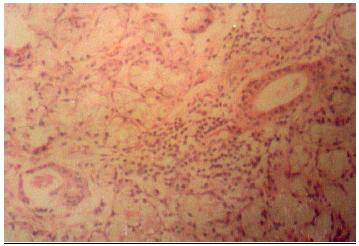
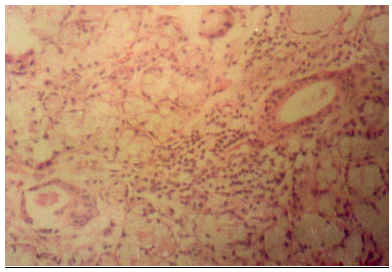
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [51.] Mpl/Fragment 081 01 - Diskussion Bearbeitet: 6. December 2014, 18:03 Singulus Erstellt: 9. November 2014, 13:53 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 81, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 90, 91, Zeilen: 90: 4 ss. ; 91: 1 ss. |
|---|---|
| [Estudios posteriores demuestran que los criterios de la CE tienen] una alta sensibilidad y una buena especificidad, en ocasiones superiores a los de San Diego o San Francisco (199).
En la actualidad se están realizando estudios para aunar los criterios europeos y americanos. El cambio más importante con respecto a los criterios europeos es que debería existir de forma imprescindible una biopsia de glándula salival patológica y/o anti-Ro/La positivos (200). De todas formas, la obligatoriedad de estos criterios debe ser tratada con cautela, debido a la gran heterogeneidad del SS en su presentación clínica. La biopsia salival es una prueba diagnóstica molesta para el paciente con sequedad oral, y en ocasiones puede no aportar el diagnóstico. Diversos autores han demostrado que la presencia de ciertos marcadores inmunológicos (IgG elevada, anti-Ro o La) pueden predecir un resultado positivo (201,202). Además, estudios recientes han demostrado alteraciones en la evaluación histológica en ciertos grupos de pacientes, como fumadores (203) o aquellos en tratamiento corticoideo (204). Respecto a la obligatoriedad de restringir el criterio inmunológico a la presencia de Ro/La, es un hecho que elimina la posibilidad de diagnosticar de SS a ciertos subgrupos epidemiológicos que precisamente se caracterizan por una pobre expresión inmunológica, como los varones (201,205), ancianos (184,206,207) o pacientes con enfermedad limitada a las mucosas (97). Además, algunos pacientes pueden presentar anticuerpos anti-Ro/La no detectables por técnicas convencionales, como IgA anti-Ro/La (208), o incluso fluctuación de los niveles de los anticuerpos a lo largo de la evolución de la enfermedad. Posiblemente, los pacientes Ro/La positivos son el subgrupo de pacientes con una presentación más activa de la enfermedad. |
Estudios posteriores demuestran que los criterios de la CE tienen una alta sensibilidad y una buena especificidad, en ocasiones superiores a los de San Diego o San Francisco (199).
[...] En la actualidad se están realizando estudios para aunar los criterios europeos y americanos. El cambio más importante con respecto a los criterios europeos es que debería existir de forma imprescindible una biopsia de glándula [página 91] salival patológica y/o anti-Ro/La positivos (200). De todas formas, la obligatoriedad de estos criterios debe ser tratada con cautela, debido a la gran heterogeneidad del SS en su presentación clínica. La biopsia salival es una prueba diagnóstica molesta para el paciente con sequedad oral, y en ocasiones puede no aportar el diagnóstico. Diversos autores han demostrado que la presencia de ciertos marcadores inmunológicos (IgG elevada, anti-Ro o La) pueden predecir un resultado positivo (201,202). Además, estudios recientes han demostrado alteraciones en la evaluación histológica en ciertos grupos de pacientes, como fumadores (203) o aquellos en tratamiento corticoideo (204). Respecto a la obligatoriedad de restringir el criterio inmunológico a la presencia de Ro/La, es un hecho que elimina la posibilidad de diagnosticar de SS a ciertos subgrupos epidemiológicos que precisamente se caracterizan por una pobre expresión inmunológica, como los varones (201,205), ancianos (184,206,207) o pacientes con enfermedad limitada a las mucosas (97). Además, algunos pacientes pueden presentar anticuerpos anti-Ro/La no detectables por técnicas convencionales, como IgA anti-Ro/La (208), o incluso fluctuación de los niveles de los anticuerpos a lo largo de la evolución de la enfermedad. Posiblemente, los pacientes Ro/La positivos son el subgrupo de pacientes con una presentación más activa de la enfermedad. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [52.] Mpl/Fragment 082 01 - Diskussion Bearbeitet: 6. December 2014, 20:17 Singulus Erstellt: 9. November 2014, 13:56 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 82, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 90, 91, Zeilen: 90: tabla; 91: 20 ss. ; 92: 1 ss. |
|---|---|
| TABLA 13. Criterios para la clasificación del SSp (Ref.88)
El carácter sindrómico del SSp y su tendencia a la evolución crónica dificultan su diagnóstico en el momento puntual en el que visitamos al paciente, y el resultado de las distintas pruebas diagnósticas varía en función del tiempo de evolución del síndrome. Fundamentalmente, la estrategia diagnóstica se basa en el estudio de los componentes ocular y bucal. Ante la sospecha de un SS una estrategia diagnóstica adecuada sería practicar primero la tinción con rosa de bengala, posteriormente la gammagrafía salival y en último término la biopsia labial (209). El diagnóstico diferencial debe realizarse con otras entidades que puedan infiltrar las glándulas salivales, fundamentalmente sarcoidosis, amiloidosis primaria y procesos linfoproliferativos (210). |
TABLA 13. Criterios para la clasificación del SSp (Ref.88)
[página 91] El carácter sindrómico del SSp y su tendencia a la evolución crónica dificultan su diagnóstico en el momento puntual en el que visitamos al paciente, y el resultado de las distintas pruebas diagnósticas varía en función del tiempo de evolución del síndrome. Fundamentalmente, la estrategia diagnóstica se basa en el estudio de los componentes ocular y bucal. Ante la sospecha de un SS una estrategia diagnóstica adecuada sería practicar primero la tinción con rosa de [página 92] bengala, posteriormente la gammagrafía salival y en último término la biopsia labial (209). El diagnóstico diferencial debe realizarse con otras entidades que puedan infiltrar las glándulas salivales, fundamentalmente sarcoidosis, amiloidosis primaria y procesos linfoproliferativos (210). |

Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [53.] Mpl/Fragment 083 01 - Diskussion Bearbeitet: 6. December 2014, 20:21 Singulus Erstellt: 9. November 2014, 14:07 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 83, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 92, 96, 97, Zeilen: 92: 5 ss. ; 96: 1; 97: 6 ss. |
|---|---|
| [Los pacientes con infecciones víricas crónicas, como la infección] por el VIH-1 o el VHC, pueden presentar un cuadro clínico, inmunológico e histológico similar (211). En los pacientes con VIH y síndrome seco, la infiltración linfocitaria de las glándulas salivales está compuesta por linfocitos CD8 positivos. En los pacientes VHC, se observa un patrón histológico muy similar, aunque a nivel inmunológico presentan negatividad de los anticuerpos anti-Ro/SSA y anti-La/SSB, así como una elevada frecuencia de crioglobulinas e hipocomplementemia.
1.7. Evolución y pronóstico del paciente con SSp 1.7.1 Desarrollo de procesos linfoproliferativos La incidencia de síndromes linfoproliferativos malignos en el SS es la más elevada entre las enfermedades autoinmunes (224) e incluso se le ha llegado a considerar como el “crossroad” entre la autoinmunidad y la proliferación linfoide maligna. Desde un punto de vista etiopatogénico, la transición de una proliferación autoinmune benigna una a transformación maligna, representa un proceso de múltiples etapas que se inicia con la activación policlonal de los linfocitos B y que culmina con una proliferación oligoclonal/monoclonal, seleccionándose así una subpoblación específica de linfocitos B. De hecho, la mayoría de los linfomas observados en los pacientes con SS son de origen linfocitario B, a pesar de que la mayoría de las células que infiltran las glándulas salivales son linfocitos T. La relación entre linfoma y SS se conoce desde 1951, cuando Rothman et al (225) describieron el primer caso. En 1964, Talal y Bunim (11) publicaron el primer estudio sobre la incidencia de linfoma en una cohorte de 58 pacientes con SS seguidos durante 4 años, y observaron 3 casos de reticulosarcoma y uno de macroglobulinemia [IgM.] |
Los pacientes con infecciones víricas crónicas, como la infección por el VIH-1 o el VHC, pueden presentar un cuadro clínico, inmunológico e histológico similar (211). En los pacientes con VIH y síndrome seco, la infiltración linfocitaria de las glándulas salivales está compuesta por linfocitos CD8 positivos. En los pacientes VHC, se observa un patrón histológico muy similar, aunque a nivel inmunológico presentan negatividad de los anticuerpos anti-Ro/SSA y anti-La/SSB, así como una elevada frecuencia de crioglobulinas e hipocomplementemia.
[página 96] 2. EVOLUCIÓN Y PRONÓSTICO DEL PACIENTE CON SSP [página 97] 2.2. Desarrollo de procesos linfoproliferativos La incidencia de síndromes linfoproliferativos malignos en el SS es la más elevada entre las enfermedades autoinmunes (224) e incluso se le ha llegado a considerar como el “crossroad” entre la autoinmunidad y la proliferación linfoide maligna. Desde un punto de vista etiopatogénico, la transición de una proliferación autoinmune benigna una a transformación maligna, representa un proceso de múltiples etapas que se inicia con la activación policlonal de los linfocitos B y que culmina con una proliferación oligoclonal/monoclonal, seleccionándose así una subpoblación específica de linfocitos B. De hecho, la mayoría de los linfomas observados en los pacientes con SS son de origen linfocitario B, a pesar de que la mayoría de las células que infiltran las glándulas salivales son linfocitos T. La relación entre linfoma y SS se conoce desde 1951, cuando Rothman et al (225) describieron el primer caso. En 1964, Talal y Bunim (11) publicaron el primer estudio sobre la incidencia de linfoma en una cohorte de 58 pacientes con SS seguidos durante 4 años, y observaron 3 casos de reticulosarcoma y uno de macroglobulinemia IgM. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [54.] Mpl/Fragment 084 01 - Diskussion Bearbeitet: 6. December 2014, 22:07 Singulus Erstellt: 9. November 2014, 14:09 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 84, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 97, 98, 99, Zeilen: 97: 22 ss. ; 98: 1 ss. ; 99: 1 |
|---|---|
| En 1966, Hornbaker et al (226) describieron la asociación entre el SS y reticulosarcoma nodular en un paciente que falleció debido a una crisis hemolítica. Un año después, Millar et al (227) estudiaron la presencia de enfermedades autoinmunes en 264 pacientes con linfoma y encontraron 14 casos de enfermedades autoinmunes, entre ellos, un paciente que desarrolló reticulosarcoma 4 años después del diagnóstico de SS.
En los años 70, aparecieron varios estudios centrados en la descripción de los marcadores clínicos e inmunológicos asociados con el desarrollo de malignidad. En 1972, Anderson y Talal (228) publicaron la primera recopilación de pacientes con SS y alteraciones linfoproliferativas, describiendo un total de 38 pacientes con pseudolinfoma, linfoma histiocítico, linfoma linfocítico nodular y difuso, timoma y macroglobulinemia de Waldenström (MW). Por otra parte, Hughes y Whaley (229) estudiaron la asociación entre SS y varias alteraciones linfoproliferativas (sarcoma reticulocelular, linfosarcoma, MW, pseudolinfoma) y destacaron que la existencia de una proliferación linfoide “benigna”, dentro del amplio espectro del SS, puede ser difícil de distinguir de un linfoma maligno. Pérez-Peña et al (230) publicaron otra revisión similar en 1974. En 1978, Faguet et al (231) reportaron el primer estudio sobre marcadores inmunológicos en un paciente con linfoma maligno detectado 22 meses después del diagnóstico de SS. Los autores establecieron el origen monoclonal de la proliferación, al demostrar la expansión de IgMκ en más del 90% de los linfocitos B obtenidos de los ganglios linfáticos y de la biopsia del nódulo linfoide pulmonar. El mismo año, Zulman et al (232) demostraron que 6 de los 9 linfomas que se desarrollaron en pacientes con SS, mostraban proliferación de linfocitos B productores de inmunoglobulinas monoclonales. Por otra parte, Kassan et al (233) publicaron un estudio epidemiológico que pretendió estimar el riesgo de aparición de linfoma en los pacientes con SS y, por otra parte, estudiar las características clínicas e inmunológicas [asociadas con su aparición durante el curso de la enfermedad.] |
En 1966, Hornbaker et al (226) describieron la asociación entre el SS y reticulosarcoma nodular en un paciente que falleció debido a una crisis hemolítica. Un año después, Millar et al (227) estudiaron la
[página 98] presencia de enfermedades autoinmunes en 264 pacientes con linfoma y encontraron 14 casos de enfermedades autoinmunes, entre ellos, un paciente que desarrolló reticulosarcoma 4 años después del diagnóstico de SS. En los años 70, aparecieron varios estudios centrados en la descripción de los marcadores clínicos e inmunológicos asociados con el desarrollo de malignidad. En 1972, Anderson y Talal (228) publicaron la primera recopilación de pacientes con SS y alteraciones linfoproliferativas, describiendo un total de 38 pacientes con pseudolinfoma, linfoma histiocítico, linfoma linfocítico nodular y difuso, timoma y macroglobulinemia de Waldenström (MW). Por otra parte, Hughes y Whaley (229) estudiaron la asociación entre SS y varias alteraciones linfoproliferativas (sarcoma reticulocelular, linfosarcoma, MW, pseudolinfoma) y destacaron que la existencia de una proliferación linfoide “benigna”, dentro del amplio espectro del SS, puede ser difícil de distinguir de un linfoma maligno. Pérez-Peña et al (230) publicaron otra revisión similar en 1974. En 1978, Faguet et al (231) reportaron el primer estudio sobre marcadores inmunológicos en un paciente con linfoma maligno detectado 22 meses después del diagnóstico de SS. Los autores establecieron el origen monoclonal de la proliferación, al demostrar la expansión de IgMκ en más del 90% de los linfocitos B obtenidos de los ganglios linfáticos y de la biopsia del nódulo linfoide pulmonar. El mismo año, Zulman et al (232) demostraron que 6 de los 9 linfomas que se desarrollaron en pacientes con SS, mostraban proliferación de linfocitos B productores de inmunoglobulinas monoclonales. Por otra parte, Kassan et al (233) publicaron un estudio epidemiológico que pretendió estimar el riesgo de aparición de linfoma en los pacientes con SS y, por otra parte, estudiar las características clínicas e inmunológicas asociadas con su aparición [página 99] durante el curso de la enfermedad. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [55.] Mpl/Fragment 085 01 - Diskussion Bearbeitet: 7. December 2014, 12:00 Singulus Erstellt: 9. November 2014, 16:16 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 85, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 99, 100, Zeilen: 99: 1ss.; 100: 1 ss. |
|---|---|
| Los autores estudiaron 136 mujeres con SS y encontraron que 7 pacientes desarrollaron un tipo de linfoma no-hodgkiniano (LNH) entre 6 meses y 13 años de la evolución de la enfermedad. Los autores demostraron que los pacientes con SS tenían un riesgo 44 veces mayor de desarrollar un linfoma, comparado con mujeres de la población general y que los pacientes que presentaban parotidomegalia, esplenomegalia y linfadenopatía, tenían un riesgo todavía mayor en desarrollar un linfoma. Los autores concluyeron que estas manifestaciones clínicas identifican un subgrupo de pacientes con SS que presentan una marcada reactividad linfoidea y que presentarán un riesgo más elevado en desarrollar un linfoma. Hasta ahora, este estudio se ha mantenido vigente como el principal punto de referencia sobre la elevada incidencia de linfoma en los pacientes con SS.
En los años 80, varios estudios demostraron que los pacientes con SS, además de presentar una activación policlonal persistente de los linfocitos B (demostrado por la producción de autoanticuerpos), pueden expresar igualmente un proceso monoclonal/oligoclonal. Esto se demostró por la presencia de inmunoglobulinas monoclonales y de crioglobulinas monoclonales de tipo II en suero y por la detección de cadenas monoclonales ligeras en orina. La presencia de inmunoglobulinas monoclonales y crioglobulinas se detectó principalmente en pacientes con enfermedad sistémica (extraglandular) y en general, mucho antes de que se presentara cualquier manifestación clínica sugestiva de malignidad linfoidea. Finalmente, los estudios más recientes se han enfocado en el estudio de la etiopatogenia de la proliferación linfoide en el SS, incluyendo el papel etiológico de algunos agentes infecciosos como el VEB o el VHC (como factores responsables de la expansión clonal de los linfocitos B) y el papel crucial del análisis molecular de la clonalidad de los linfocitos B (como el primer paso para el diagnóstico de malignidad). |
Los autores estudiaron 136 mujeres con SS y encontraron que 7 pacientes desarrollaron un tipo de linfoma no-hodgkiniano (LNH) entre 6 meses y 13 años de la evolución de la enfermedad. Los autores demostraron que los pacientes con SS tenían un riesgo 44 veces mayor de desarrollar un linfoma, comparado con mujeres de la población general y que los pacientes que presentaban parotidomegalia, esplenomegalia y linfadenopatía, tenían un riesgo todavía mayor en desarrollar un linfoma. Los autores concluyeron que estas manifestaciones clínicas identifican un subgrupo de pacientes con SS que presentan una marcada reactividad linfoidea y que presentarán un riesgo más elevado en desarrollar un linfoma. Hasta ahora, este estudio se ha mantenido vigente como el principal punto de referencia sobre la elevada incidencia de linfoma en los pacientes con SS.
En los años 80, varios estudios demostraron que los pacientes con SS, además de presentar una activación policlonal persistente de los linfocitos B (demostrado por la producción de autoanticuerpos), pueden expresar igualmente un proceso monoclonal/oligoclonal. Esto se demostró por la presencia de inmunoglobulinas monoclonales y de crioglobulinas monoclonales de tipo II en suero y por la detección de cadenas monoclonales ligeras en orina. La presencia de inmunoglobulinas monoclonales y crioglobulinas se detectó principalmente en pacientes con enfermedad sistémica (extraglandular) y en general, mucho antes de que se presentara cualquier manifestación clínica sugestiva de malignidad linfoidea. Finalmente, los estudios más recientes se han enfocado en el estudio de la etiopatogenia de la proliferación linfoide en el SS, incluyendo el papel etiológico de algunos agentes infecciosos como el VEB o el VHC (como [página 100] factores responsables de la expansión clonal de los linfocitos B) y el papel crucial del análisis molecular de la clonalidad de los linfocitos B (como el primer paso para el diagnóstico de malignidad). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [56.] Mpl/Fragment 086 01 - Diskussion Bearbeitet: 7. December 2014, 12:04 Singulus Erstellt: 9. November 2014, 16:18 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 86, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 100, Zeilen: 4 ss. |
|---|---|
| 1.7.1.1. Estudios epidemiológicos
Tal como ya se comentó, en 1978 se publicó el primer estudio prospectivo sobre la incidencia de linfoma en pacientes con SS. Kassan et al (233) encontraron que el riesgo de aparición alcanzaba los 6.4 casos por 1000 habitantes por año (44 veces más que la población general). Desde entonces, la mayoría de los estudios analizan retrospectivamente la incidencia de linfoma en pacientes con SS, encontrando porcentajes que oscilan entre el 1-10% (11,12,116,126,132,188,193,233-245) (Tabla 14). TABLA 14. Prevalencia de linfoma en el SSp según diferentes autores
|
2.2.1. Estudios epidemiológicos
Tal como ya se comentó, en 1978 se publicó el primer estudio prospectivo sobre la incidencia de linfoma en pacientes con SS. Kassan et al (233) encontraron que el riesgo de aparición alcanzaba los 6.4 casos por 1000 habitantes por año (44 veces más que la población general). Desde entonces, la mayoría de los estudios analizan retrospectivamente la incidencia de linfoma en pacientes con SS, encontrando porcentajes que oscilan entre el 1-10% (11,12,116,126,132,188,193,233-245) (Tabla 14). TABLA 14. Prevalencia de linfoma en el SSp según diferentes autores
|
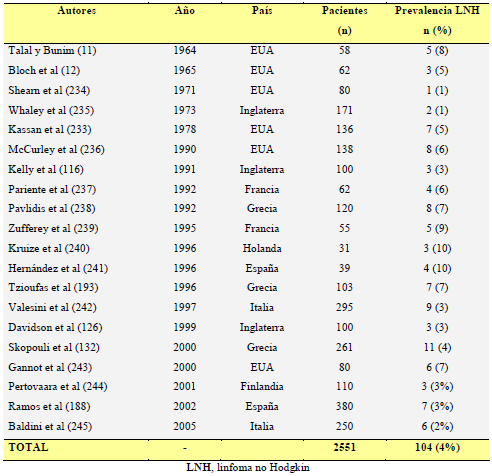
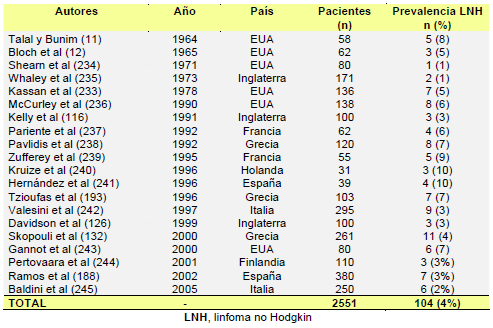
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [57.] Mpl/Fragment 087 01 - Diskussion Bearbeitet: 7. December 2014, 12:08 Singulus Erstellt: 9. November 2014, 16:20 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 87, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 101, 102, Zeilen: 101: 1 ss.; 102: 1 ss. |
|---|---|
| La diferencia entre las cifras obtenidas se debe, por un lado, a los distintos criterios diagnósticos del SS, y por otro, al tiempo de seguimiento de la enfermedad.
La mayoría de los estudios muestran que los pacientes con SSp tienen un riesgo más elevado en desarrollar enfermedades linfoproliferativas que los pacientes con síndrome seco o SS asociado. Kruize et al (240) no encontraron enfermedad linfoproliferativa en aquellos pacientes que presentaban síntomas de queratoconjuntivitis ó SS asociado a otra enfermedad autoinmune. En otro estudio similar, en 331 pacientes italianos (242), no se detectó linfoma en ningún paciente con SS asociado. Por otra parte, un estudio epidemiológico reciente en pacientes finlandeses (246) demostró una mayor incidencia de LNH en el SSp, con un riesgo relativo de 8.7, comparado con el 4.5 y 2.2 para el SS asociado y la AR, respectivamente. Finalmente, se ha descrito que aquellos pacientes con aparición precoz del SS tienen un riesgo más elevado de presentar procesos linfoproliferativos. En el estudio de Kassan et al (233), los pacientes que desarrollaron SS antes de los 45 años de edad, presentaban un riesgo 60 veces superior de desarrollar linfoma comparado con la población general. Otros estudios (247), han demostrado una mayor incidencia de linfadenopatías, FR, inmunoglobulinas monoclonales y linfoma en pacientes cuya enfermedad apareció antes de los 35 años de edad. 1.7.1.2. Etiopatogenia a) Alteraciones genéticas Las traslocaciones cromosómicas son las alteraciones genéticas más frecuentes asociadas a la linfomagénesis de los linfocitos B. Estos cambios originan una alteración funcional en los diversos genes implicados en el control del ciclo celular y de los fenómenos de diferenciación y muerte celular programada. |
La diferencia entre las cifras obtenidas se debe, por un lado, a los distintos criterios diagnósticos del SS, y por otro, al tiempo de seguimiento de la enfermedad.
La mayoría de los estudios muestran que los pacientes con SSp tienen un riesgo más elevado en desarrollar enfermedades linfoproliferativas que los pacientes con síndrome seco o SS asociado. Kruize et al (240) no encontraron enfermedad linfoproliferativa en aquellos pacientes que presentaban síntomas de queratoconjuntivitis ó SS asociado a otra enfermedad autoinmune. En otro estudio similar, en 331 pacientes italianos (242), no se detectó linfoma en ningún paciente con SS asociado. Por otra parte, un estudio epidemiológico reciente en pacientes finlandeses (246) demostró una mayor incidencia de LNH en el SSp, con un riesgo relativo de 8.7, comparado con el 4.5 y 2.2 para el SS asociado y la AR, respectivamente. Finalmente, se ha descrito que aquellos pacientes con aparición precoz del SS tienen un riesgo más elevado de presentar procesos linfoproliferativos. En el estudio de Kassan et al (233), los pacientes que desarrollaron SS antes de los 45 años de edad, presentaban un riesgo 60 veces superior de desarrollar linfoma comparado con la población general. Otros estudios (247), han demostrado una mayor incidencia de linfadenopatías, FR, inmunoglobulinas monoclonales y linfoma en pacientes cuya enfermedad apareció antes de los 35 años de edad. 2.2.2. Etiopatogenia a) Alteraciones genéticas Las traslocaciones cromosómicas son las alteraciones genéticas más frecuentes asociadas a la linfomagénesis de los linfocitos B. Estos cambios [página 102] originan una alteración funcional en los diversos genes implicados en el control del ciclo celular y de los fenómenos de diferenciación y muerte celular programada. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [58.] Mpl/Fragment 088 01 - Diskussion Bearbeitet: 7. December 2014, 14:55 Singulus Erstellt: 9. November 2014, 16:22 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 88, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 102, 103, Zeilen: 102: 3 ss.; 103: 1 ss. |
|---|---|
| [Es importante mencionar] que además de los hallazgos morfológicos e inmunofenotípicos característicos en la mayoría de los síndromes linfoproliferativos de origen B, se han descrito alteraciones genéticas y moleculares propias de cada tipo de linfoma. Esto se debe a que los diferentes síndromes linfoproliferativos B provienen de la transformación neoplásica de los linfocitos B en sus diferentes etapas de maduración. La gran heterogeneidad, tanto biológica como clínica, de los linfomas se fundamenta en su origen y, por tanto, en las diferentes características genéticas y moleculares de los mismos (248).
Se han descrito varias alteraciones cromosómicas en los linfomas de pacientes con SS. Una de ellas es la traslocación t(14;18), un hallazgo citogenético característico del linfoma folicular (LF). Esta traslocación ocasiona la yuxtaposición del gen bcl2 junto al gen de la cadena pesada de las inmunoglobulinas, lo que motiva una producción muy elevada de la proteína bcl2 (249). Aunque aún no está totalmente esclarecida la función de la proteína bcl2, se sabe que tiene un gran efecto anti-apoptótico, esto es, efecto inhibitorio de la muerte celular programada. Esto conduce a un aumento de la supervivencia de las células linfoides y por consiguiente, a un mayor riesgo de transformación neoplásica. Algunos autores han estudiado la presencia de esta traslocación en pacientes con SS y linfoma. Fox et al (250) han demostrado la presencia de la translocación del gen bcl2 en el 50% de los linfomas asociados al SS, más no se detectó en aquellos pacientes con SS sin linfoma. Pisa et al (251) también observaron la traslocación del gen bcl2 en 5 de 7 pacientes con SS y linfoma asociado, más no encontraron esta traslocación en 50 biopsias de las glándulas salivales de pacientes con SS que no presentaban clínica de linfoma, ni en las biopsias del mismo paciente, previas a la aparición del linfoma. Recientemente, se han estudiado 100 pacientes con SSp, detectándose la traslocación t(14,18) en un 5% de los pacientes, aunque en algunos casos la detección de la traslocación fue transitoria (252). |
Es importante mencionar que además de los hallazgos morfológicos e inmunofenotípicos característicos en la mayoría de los síndromes linfoproliferativos de origen B, se han descrito alteraciones genéticas y moleculares propias de cada tipo de linfoma. Esto se debe a que los diferentes síndromes linfoproliferativos B provienen de la transformación neoplásica de los linfocitos B en sus diferentes etapas de maduración. La gran heterogeneidad, tanto biológica como clínica, de los linfomas se fundamenta en su origen y, por tanto, en las diferentes características genéticas y moleculares de los mismos (248).
Se han descrito varias alteraciones cromosómicas en los linfomas de pacientes con SS. Una de ellas es la traslocación t(14;18), un hallazgo citogenético característico del linfoma folicular (FL). Esta traslocación ocasiona la yuxtaposición del gen bcl2 junto al gen de la cadena pesada de las inmunoglobulinas, lo que motiva una producción muy elevada de la proteína bcl2 (249). Aunque aún no está totalmente esclarecida la función de la proteína bcl2, se sabe que tiene un gran efecto anti-apoptótico, esto es, efecto inhibitorio de la muerte celular programada. Esto conduce a un aumento de la supervivencia de las células linfoides y por consiguiente, a un mayor riesgo de transformación neoplásica. Algunos autores han estudiado la presencia de esta traslocación en pacientes con SS y linfoma. Fox et al (250) han demostrado la presencia de la translocación del gen bcl2 en el 50% de los linfomas asociados al SS, más no se detectó en aquellos pacientes con SS sin linfoma. Pisa et al (251) también observaron la traslocación del gen bcl2 en 5 de 7 pacientes con [página 103] SS y linfoma asociado, más no encontraron esta traslocación en 50 biopsias de las glándulas salivales de pacientes con SS que no presentaban clínica de linfoma, ni en las biopsias del mismo paciente, previas a la aparición del linfoma. Recientemente, se han estudiado 100 pacientes con SSp, detectándose la traslocación t(14,18) en un 5% de los pacientes, aunque en algunos casos la detección de la traslocación fue transitoria (252). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [59.] Mpl/Fragment 089 01 - Diskussion Bearbeitet: 7. December 2014, 15:01 Singulus Erstellt: 9. November 2014, 16:24 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 89, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 103, 104, Zeilen: 103: 6ss.; 104: 1 ss. |
|---|---|
| [Por el contrario, otros autores] no han podido identificar esta traslocación, t(14;18) en biopsias labiales que mostraban monoclonolidad de las cadenas pesadas o en algunos linfomas de glándulas extrasalivales (253). Por tanto, el análisis de la traslocación del gen bcl2 en las biopsias de tejido contribuye al diagnóstico de linfoma, aunque un resultado negativo no elimina la posibilidad de una neoplasia. Se han descrito otras alteraciones genéticas en pacientes con SS y linfoma, como la presencia de mutaciones en el gen p53 (relacionado con la supresión de la actividad tumoral). Finalmente, Gou et al (254), han descrito una reparación defectuosa del DNA, con la consiguiente producción de O-metilguanina (material genético no reparado) en pacientes con SSp que desarrollaron linfoma.
b) Agentes infecciosos Ciertos agentes infecciosos, principalmente los virus, juegan un papel etiopatogénico en la aparición de linfomas. Diversos virus como el VEB y el VHC infectan directamente los linfocitos B y promueven su proliferación por la expresión de proteínas transformadoras de los virus codificados. Por otra parte, los agentes infecciosos pueden ser los causantes de determinantes antigénicos, los cuales inducen y mantienen la proliferación de los linfocitos B en caso de una infección crónica. VEB. El VEB, es el herpes virus causante de la mononucleosis infecciosa y directamente relacionado con el linfoma de Burkitt, el carcinoma nasofaríngeo y el síndrome linfoproliferativo asociado al cromosoma X. Es capaz de replicarse en las glándulas salivales y el epitelio nasofaríngeo durante la infección primaria y persistir latente por el resto de la vida en el huésped (255). Además, tiene la capacidad de infectar y producir hiperactividad linfoide B y promover así la síntesis policlonal de las inmunoglobulinas (256). |
Por el contrario, otros autores no han podido identificar esta traslocación, t(14;18) en biopsias labiales que mostraban monoclonolidad de las cadenas pesadas o en algunos linfomas de glándulas extrasalivales (253). Por tanto, el análisis de la traslocación del gen bcl2 en las biopsias de tejido contribuye al diagnóstico de linfoma, aunque un resultado negativo no elimina la posibilidad de una neoplasia. Se han descrito otras alteraciones genéticas en pacientes con SS y linfoma, como la presencia de mutaciones en el gen p53 (relacionado con la supresión de la actividad tumoral. Finalmente, Gou et al (254), han descrito una reparación defectuosa del DNA, con la consiguiente producción de O-metilguanina (material genético no reparado) en pacientes con SSp que desarrollaron linfoma.
b) Agentes infecciosos Ciertos agentes infecciosos, principalmente los virus, juegan un papel etiopatogénico en la aparición de linfomas. Diversos virus como el VEB y el VHC infectan directamente los linfocitos B y promueven su proliferación por la expresión de proteínas transformadoras de los virus codificados. Por otra parte, los agentes infecciosos pueden ser los causantes de determinantes [página 104] antigénicos, los cuales inducen y mantienen la proliferación de los linfocitos B en caso de una infección crónica. VEB. El VEB, es el herpes virus causante de la mononucleosis infecciosa y directamente relacionado con el linfoma de Burkitt, el carcinoma nasofaríngeo y el síndrome linfoproliferativo asociado al cromosoma X. Es capaz de replicarse en las glándulas salivales y el epitelio nasofaríngeo durante la infección primaria y persistir latente por el resto de la vida en el huésped (255). Además, tiene la capacidad de infectar y producir hiperactividad linfoide B y promover así la síntesis policlonal de las inmunoglobulinas (256). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [60.] Mpl/Fragment 090 01 - Diskussion Bearbeitet: 7. December 2014, 14:59 Singulus Erstellt: 9. November 2014, 16:28 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 90, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 104, 105, Zeilen: 104: 9 ss.; 105: 1 ss. |
|---|---|
| [Hay evidencia de que el VEB juega un papel importante en la] linfomagénesis observada en pacientes con SS. Los genes del VEB y/o la expresión de proteínas en las biopsias de las glándulas salivales de estos pacientes es elevada comparada con la del grupo control (257,258). Otros autores han encontrado niveles elevados de DNA del VEB en la saliva de pacientes con SS y pseudolinfoma (258). Por otra parte, hay autores que han detectado el VEB en tejido linfomatoso: Fox et al (259) encontraron el DNA del VEB en 2 de 5 adenopatías cervicales de pacientes con SS y LNH, Jeffers et al (260) lo encontraron en 3 de 6 pacientes con SS y linfoma MALT asociado, Royer et al (261) en 1 de 4 linfomas parotídeos y por último, Freimark et al (262) en 1 de 9 linfomas. Sin embargo, hay autores que no han encontrado evidencia del VEB en el linfoma de bajo grado en pacientes con SS (260). El VEB se encuentra latente en el epitelio ductal de las glándulas salivales y lacrimales, donde puede reactivarse por posibles defectos en la regulación de los linfocitos B, como las mutaciones del VEB que escaparían al reconocimiento de las células citotóxicas específicas del virus (259). Por otra parte, el epitelio de las glándulas salivales de los pacientes con SS expresan niveles elevados de antígenos HLA-DR, por lo que tienen capacidad de presentar los antígenos del VEB. El SS puede representar una situación, en la cual individuos genéticamente predispuestos (por ejemplo, HLA-DR3-DQA4-DQB2) tienen una persistente pero ineficaz respuesta inmune contra el VEB en el tejido glandular (259).
VHH-6. El reciente aislamiento de un nuevo miembro de la familia del herpes virus (HHV-6) en pacientes con enfermedades linfoproliferativas, ha llevado a varios autores a estudiar este virus en varias muestras tisulares (259) y salivales (263) de pacientes con SSp que desarrollaron un LNH. Jarrett et al (264) analizaron muestras de tejido de un paciente con linfoma que se desarrolló en el contexto del SS, y encontraron [secuencias de DNA, específicos del HHV-6.] |
Hay evidencia de que el VEB juega un papel importante en la linfomagénesis observada en pacientes con SS. Los genes del VEB y/o la expresión de proteínas en las biopsias de las glándulas salivales de estos pacientes es elevada comparada con la del grupo control (257,258). Otros autores han encontrado niveles elevados de DNA del VEB en la saliva de pacientes con SS y pseudolinfoma (258). Por otra parte, hay autores que han detectado el VEB en tejido linfomatoso: Fox et al (259) encontraron el DNA del VEB en 2 de 5 adenopatías cervicales de pacientes con SS y LNH, Jeffers et al (260) lo encontraron en 3 de 6 pacientes con SS y linfoma MALT asociado, Royer et al (261) en 1 de 4 linfomas parotídeos y por último, Freimark et al (262) en 1 de 9 linfomas. Sin embargo, hay autores que no han encontrado evidencia del VEB en el linfoma de bajo grado en pacientes con SS (260). El VEB se encuentra latente en el epitelio ductal de las glándulas salivales y lacrimales, donde puede reactivarse por posibles defectos en la regulación de los linfocitos B, como las mutaciones del VEB que escaparían al reconocimiento de las células
[página 105] citotóxicas específicas del virus (259). Por otra parte, el epitelio de las glándulas salivales de los pacientes con SS expresan niveles elevados de antígenos HLA-DR, por lo que tienen capacidad de presentar los antígenos del VEB. El SS puede representar una situación, en la cual individuos genéticamente predispuestos (por ejemplo, HLA-DR3-DQA4-DQB2) tienen una persistente pero ineficaz respuesta inmune contra el VEB en el tejido glandular (259). VHH-6. El reciente aislamiento de un nuevo miembro de la familia del herpes virus (HHV-6) en pacientes con enfermedades linfoproliferativas, ha llevado a varios autores a estudiar este virus en varias muestras tisulares (259) y salivales (263) de pacientes con SSp que desarrollaron un LNH. Jarrett et al (264) analizaron muestras de tejido de un paciente con linfoma que se desarrolló en el contexto del SS, y encontraron secuencias de DNA, específicos del HHV-6. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [61.] Mpl/Fragment 091 01 - Diskussion Bearbeitet: 7. December 2014, 16:30 Singulus Erstellt: 9. November 2014, 16:31 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 91, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 105, 106, Zeilen: 105: 14 ss.; 105: 1 ss. |
|---|---|
| Igualmente, Fox et al (265) encontraron secuencias de DNA del HHV-6 en ganglios linfáticos de 1 de 14 pacientes.
VHC. En 1992, Haddad et al (266) postularon una posible relación entre el VHC (un virus que puede excretarse por la saliva) y el SS. Estos autores describieron la aparición de cambios histológicos característicos del SS en las glándulas salivales de pacientes con infección por el VHC. Recientemente, dos estudios clínicos, que incluyen grandes series de pacientes, describieron las características clínicas e inmunológicas de este subgrupo de pacientes con SS e infección por el VHC (267-268). Además, Koike et al, (269) publicaron la primera evidencia experimental de la posible relación entre el SS y la infección por el VHC. Estos autores encontraron una exocrinopatía similar al SS en las glándulas salivales y lacrimales de ratones transgénicos para determinados genes del VHC. Por otra parte, la asociación con la proliferación linfoide se demostró en años recientes. En 1971, Heimann (270) describió la frecuente asociación entre la cirrosis hepática y las enfermedades linfoproliferativas, destacando el posible papel de los virus hepatotropos en la patogenia de ambas enfermedades. Veintitrés años después, en 1994, se demostró la replicación del VHC en suero y linfocitos periféricos en un tercio de pacientes italianos con LNH (271,272). Estudios posteriores confirmaron la elevada incidencia de infección por el VHC en el LNH (273,274). Franzin et al (275) encontraron una elevada frecuencia de expansión clonal de linfocitos B en pacientes infectados por el VHC en ausencia de crioglobulinemia. Finalmente, se han realizado estudios que han detectado el VHC en el tejido linfomatoso (276,277). De Vita et al (278) describieron los linfomas B en 35 pacientes con VHC y encontraron que la CM ó el SS precedía a la aparición de LNH en 5 pacientes y al comparar los sitios de afección por el LNH, descubrieron que la afección hepática y [salival era más frecuentes en pacientes VHC positivos comparados con pacientes VHC negativos.] |
Igualmente, Fox et al (265) encontraron secuencias de DNA del HHV-6 en ganglios linfáticos de 1 de 14 pacientes.
VHC. En 1992, Haddad et al (266) postularon una posible relación entre el VHC (un virus que puede excretarse por la saliva) y el SS. Estos autores describieron la aparición de cambios histológicos característicos del SS en las glándulas salivales de pacientes con infección por el VHC. Recientemente, dos estudios clínicos, que incluyen grandes series de pacientes, describieron las características clínicas e inmunológicas de este subgrupo de pacientes con SS e infección por el VHC (267-268). Además, Koike et al, (269) publicaron la primera evidencia experimental de la posible relación entre el SS y la infección [página 106] por el VHC. Estos autores encontraron una exocrinopatía similar al SS en las glándulas salivales y lacrimales de ratones transgénicos para determinados genes del VHC. Por otra parte, la asociación con la proliferación linfoide se demostró en años recientes. En 1971, Heimann (270) describió la frecuente asociación entre la cirrosis hepática y las enfermedades linfoproliferativas, destacando el posible papel de los virus hepatotropos en la patogenia de ambas enfermedades. Veintitrés años después, en 1994, se demostró la replicación del VHC en suero y linfocitos periféricos en un tercio de pacientes italianos con LNH (271,272). Estudios posteriores confirmaron la elevada incidencia de infección por el VHC en el LNH (273,274). Franzin et al (275) encontraron una elevada frecuencia de expansión clonal de linfocitos B en pacientes infectados por el VHC en ausencia de crioglobulinemia. Finalmente, se han realizado estudios que han detectado el VHC en el tejido linfomatoso (276,277). De Vita et al (278) describieron los linfomas B en 35 pacientes con VHC y encontraron que la CM ó el SS precedía a la aparición de LNH en 5 pacientes y al comparar los sitios de afección por el LNH, descubrieron que la afección hepática y salival era más frecuentes en pacientes VHC positivos comparados con pacientes VHC negativos. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [62.] Mpl/Fragment 092 01 - Diskussion Bearbeitet: 7. December 2014, 16:33 Singulus Erstellt: 9. November 2014, 21:24 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 92, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 106, 107, Zeilen: 106: 15 ss.; 107: 1 ss. |
|---|---|
| [De Vita et al (278) describieron los linfomas B en 35 pacientes con VHC y encontraron que la CM ó el SS precedía a la aparición de LNH en 5 pacientes y al comparar los sitios de afección por el LNH, descubrieron que la afección hepática y] salival era más frecuentes en pacientes VHC positivos comparados con pacientes VHC negativos. El VHC muestra un especial tropismo por el hígado y las glándulas salivales mayores, su afección parece ser mayor en pacientes VHC positivos con LNH, mientras que la localización en estos órganos es extremadamente infrecuente en pacientes con LNH y VHC negativos (279-281). Por tanto, se puede concluir, que el VHC infecta y se replica activamente en los hepatocitos (282,283) y en el epitelio glándular salival (276), que el ácido ribonucleico (RNA) del VHC se detecta en sangre periférica de células mononucleares de pacientes con hepatitis C crónica (284), y que debido a su linfotropismo/sialotropismo y hepatotropismo, el VHC expresa su potencial oncogénico en dos direcciones diferentes: 1) a neoplasias de linfocitos B (en algunos casos, en pacientes con SS o crioglobulinemia previa) y 2) cáncer de hígado. Por lo tanto, es más apropiado considerar la infección crónica por el VHC como un síndrome clínico multisistémico más que como una simple enfermedad del hígado (284-286).
Helicobacter pylori. En estos últimos años, se ha implicado a un pequeño bacilo Gram negativo, denominado Helicobacter pylori, en la patología gastroduodenal más importante, esto es la úlcera péptica y el carcinoma gástrico. Recientes estudios epidemiológicos y experimentales, también han relacionado al H.pylori con la aparición de linfomas MALT (tejido linfoide asociado a las mucosas) gástricos. En este sentido, diferentes grupos han demostrado el papel de los linfocitos T específicos para H. pylori en la proliferación de linfomas, así como la regresión del linfoma MALT después de la erradicación del bacilo (287-290). Parece que la infección persistente por H.pylori promueve la proliferación linfocitaria, la cual puede volverse autónoma y evolucionar a una enfermedad neoplásica linfoproliferativa. Así, se ha postulado recientemente, una posible relación entre el H.pylori y la linfomagenesis gástrica en el SS. |
De Vita et al (278) describieron los linfomas B en 35 pacientes con VHC y encontraron que la CM ó el SS precedía a la aparición de LNH en 5 pacientes y al comparar los sitios de afección por el LNH, descubrieron que la afección hepática y salival era más frecuentes en pacientes VHC positivos comparados con pacientes VHC negativos. El VHC muestra un especial tropismo por el hígado y las glándulas salivales mayores, su afección parece ser mayor en pacientes VHC positivos con LNH, mientras que la localización en estos órganos es extremadamente infrecuente en pacientes con LNH y VHC negativos (279-281). Por tanto, se puede concluir, que el VHC infecta y se replica activamente en los hepatocitos (282,283) y en el epitelio glándular salival (276), que el ácido ribonucleico (RNA) del VHC se detecta en sangre
[página 107] periférica de células mononucleares de pacientes con hepatitis C crónica (284), y que debido a su linfotropismo/sialotropismo y hepatotropismo, el VHC expresa su potencial oncogénico en dos direcciones diferentes: 1) a neoplasias de linfocitos B (en algunos casos, en pacientes con SS o crioglobulinemia previa) y 2) cáncer de hígado. Por lo tanto, es más apropiado considerar la infección crónica por el VHC como un síndrome clínico multisistémico más que como una simple enfermedad del hígado (284-286). Helicobacter pylori. En estos últimos años, se ha implicado a un pequeño bacilo Gram negativo, denominado Helicobacter pylori, en la patología gastroduodenal más importante, esto es la úlcera péptica y el carcinoma gástrico. Recientes estudios epidemiológicos y experimentales, también han relacionado al H.pylori con la aparición de linfomas MALT (tejido linfoide asociado a las mucosas) gástricos. En este sentido, diferentes grupos han demostrado el papel de los linfocitos T específicos para H. pylori en la proliferación de linfomas, así como la regresión del linfoma MALT después de la erradicación del bacilo (287-290). Parece que la infección persistente por H.pylori promueve la proliferación linfocitaria, la cual puede volverse autónoma y evolucionar a una enfermedad neoplásica linfoproliferativa. Así, se ha postulado recientemente, una posible relación entre el H.pylori y la linfomagenesis gástrica en el SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [63.] Mpl/Fragment 093 01 - Diskussion Bearbeitet: 7. December 2014, 16:36 Singulus Erstellt: 9. November 2014, 21:26 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 93, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 107, 108, Zeilen: 107: 20 ss.; 108: 1 ss. |
|---|---|
| [La acumulación] linfoidea en la mucosa gástrica es un hecho frecuente en el SS, aunque aún no se ha demostrado la expansión de linfocitos B ocasionada por un antígeno específico. De Vita et al (289) describieron un linfoma gástrico de bajo grado con infección concomitante por el H.pylori en un paciente con SS. Después de la erradicación del germen, se observó la desaparición completa del linfoma gástrico aunque no hubo mejoría en la afección parotídea y nodal del SS. Múltiples análisis moleculares han demostrado la expansión de la misma clona de linfocitos B en lesiones linfoides sincrónicas y metacrónicas en lesiones parotídeas y gástricas antes y después de la erradicación del H.pylori. Por otra parte, otros autores (290) han estudiado el tejido gástrico en el SS para definir si la presencia de linfomas MALT gástricos estaban asociados a agentes infecciosos y encontraron que la infección por H.pylori no es más frecuente entre los pacientes con SS comparado con los controles, y que los linfomas MALT pueden aparecer en el estómago aún en ausencia de la infección por H. pylori. Otros estudios realizados en un número limitado de pacientes con SS y dispepsia demostraron que la clonalidad puede persistir por más de 6 meses después de la erradicación del H.pylori (291). Finalmente, se ha detectado la existencia del H. Pylori en un linfoma MALT de glándula salival en un paciente con SS, lo que destaca el papel del H. Pylori como agente desencadenante de la proliferación (292).
1.7.1.3. Clasificación histológica La actual clasificación de los linfomas corresponde a la “Clasificación Revisada de Linfoma Americano-Europea” (REAL) o al de la Organización Mundial de la Salud (OMS), que incluye más de 30 entidades clínicopatológicas. Sin embargo, no todos ellos se han relacionado con cierta frecuencia al SS. De hecho, la inmensa mayoría de los síndromes linfoproliferativos asociados al SS son de estirpe B. |
La acumulación linfoidea en la mucosa gástrica es un hecho frecuente en el SS, aunque aún no se ha demostrado la expansión de linfocitos B ocasionada por un antígeno específico. De Vita et al (289) describieron un linfoma gástrico de bajo grado con infección concomitante por el H.pylori en un paciente con SS. Después de la erradicación
[página 108] del germen, se observó la desaparición completa del linfoma gástrico aunque no hubo mejoría en la afección parotídea y nodal del SS. Múltiples análisis moleculares han demostrado la expansión de la misma clona de linfocitos B en lesiones linfoides sincrónicas y metacrónicas en lesiones parotídeas y gástricas antes y después de la erradicación del H.pylori. Por otra parte, otros autores (290) han estudiado el tejido gástrico en el SS para definir si la presencia de linfomas MALT gástricos estaban asociados a agentes infecciosos y encontraron que la infección por H.pylori no es más frecuente entre los pacientes con SS comparado con los controles, y que los linfomas MALT pueden aparecer en el estómago aún en ausencia de la infección por H. pylori. Otros estudios realizados en un número limitado de pacientes con SS y dispepsia demostraron que la clonalidad puede persistir por más de 6 meses después de la erradicación del H.pylori (291). Finalmente, se ha detectado la existencia del H. Pylori en un linfoma MALT de glándula salival en un paciente con SS, lo que destaca el papel del H. Pylori como agente desencadenante de la proliferación (292). 2.2.3. Clasificación histológica La actual clasificación de los linfomas corresponde a la “Clasificación Revisada de Linfoma Americano-Europea” (REAL) o al de la Organización Mundial de la Salud (OMS), que incluye más de 30 entidades clínicopatológicas. Sin embargo, no todos ellos se han relacionado con cierta frecuencia al SS. De hecho, la inmensa mayoría de los síndromes linfoproliferativos asociados al SS son de estirpe B. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [64.] Mpl/Fragment 094 01 - Diskussion Bearbeitet: 7. December 2014, 17:27 Singulus Erstellt: 9. November 2014, 21:29 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 94, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 108, 109, 110, Zeilen: 108: 23; 109: 1 ss. ; 110: tabla |
|---|---|
| [Las otras] enfermedades linfoproliferativas son infrecuentes y todo hace pensar que su relación con el SS podría ser más bien casual.
Por otra parte, es posible observar imágenes clínicas sugestivas de malignidad en algunos pacientes pero que no se clasifican como malignas, aún por medio de técnicas modernas de genética molecular como el inmunofenotipado y el genotipado. El término pseudolinfoma, introducido por Godwin en 1952 (293), se aplica a tales casos (294) y se considera como una etapa intermedia en la transición de una proliferación linfoide benigna a una maligna. En la descripción original de pseudolinfoma en las glándulas salivales, se encontraron pequeños linfocitos, células plasmáticas, inmunoblastos y distintas poblaciones de células mononucleares (294). En estudios posteriores se ha demostrado claramente la naturaleza B de esta población mononuclear (295-297). Actualmente, se acepta la designación de “linfocitos B monocitoides” como término apropiado para estas células. El término pseudolinfoma en la literatura antigua correspondía a la mayoría de los casos a linfomas de bajo grado. a) Síndromes linfoproliferativos B Se han descrito en la literatura varios subtipos histológicos de linfomas B en pacientes con SS (236-238,298-303) (Tabla 15). Tabla 15. Subtipos histológicos de los linfomas de células B descritos en pacientes con SSp
|
Las otras enfermedades
[página 109] linfoproliferativas son infrecuentes y todo hace pensar que su relación con el SS podría ser más bien casual. Por otra parte, es posible observar imágenes clínicas sugestivas de malignidad en algunos pacientes pero que no se clasifican como malignas, aún por medio de técnicas modernas de genética molecular como el inmunofenotipado y el genotipado. El término pseudolinfoma, introducido por Godwin en 1952 (293), se aplica a tales casos (294) y se considera como una etapa intermedia en la transición de una proliferación linfoide benigna a una maligna. En la descripción original de pseudolinfoma en las glándulas salivales, se encontraron pequeños linfocitos, células plasmáticas, inmunoblastos y distintas poblaciones de células mononucleares (294). En estudios posteriores se ha demostrado claramente la naturaleza B de esta población mononuclear (295-297). Actualmente, se acepta la designación de “linfocitos B monocitoides” como término apropiado para estas células. El término pseudolinfoma en la literatura antigua correspondía a la mayoría de los casos a linfomas de bajo grado. a) Síndromes linfoproliferativos B Se han descrito en la literatura varios subtipos histológicos de linfomas B en pacientes con SS (236-238,298-303) (Tabla 15). [página 110] Tabla 15. Subtipos histológicos de los linfomas de células B descritos en pacientes con SSp
|


Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [65.] Mpl/Fragment 095 01 - Diskussion Bearbeitet: 7. December 2014, 17:33 Singulus Erstellt: 9. November 2014, 21:30 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 95, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 110, 111, Zeilen: 110: 1 ss.; 111: 1 ss. |
|---|---|
| Estos tumores se distinguen en la mayoría de los casos por una combinación de características morfológicas e inmunofenotípicas específicas (304). El linfoma más frecuente de linfocitos B descrito en pacientes con SS es el MZL, que incluye el linfoma de bajo grado de linfocitos B tipo MALT y el MBCL (249).
La denominación MALT consiste en el tejido linfoide extranodal asociado al epitelio gastrointestinal, bronquial y tejidos de las mucosas. Está compuesto de linfocitos que pueden invadir estos lugares para procesar antígenos luminales y así proporcionar inmunidad de la mucosa. Los linfomas de bajo grado pueden surgir de los tejidos linfoides (MALT-linfomas) y del tejido linfoide asociado a otros tipos de epitelio. Los linfomas MALT se presentan con frecuencia como enfermedad extraganglionar localizada que afecta al tejido epitelial glandular. Isaacson et al (301,305) fueron los primeros en mencionar el concepto de linfomas extranodales MALT. Se ha descrito que este grupo de linfomas surge del tejido extranodal del tubo digestivo, glándulas salivales, pulmón y tiroides (301,305,306) y con poca frecuencia del tejido extranodal normal como el de las placas de Peyer. Los linfomas MALT de bajo grado se caracterizan por presentar un curso clínico indolente y una similitud en la organización del MALT normal. Los linfomas MALT poseen características poco conocidas, como el mecanismo por el cual las células neoplásicas permanecen confinadas a un solo lugar, la presencia o ausencia de tráfico neoplásico celular y la diseminación específica de linfomas MALT a otras mucosas. Por otra parte, los linfomas MALT de bajo grado pueden sufrir una transformación histológica a formas más agresivas, habitualmente a un linfoma difuso de células grandes, con el consiguiente cambio en el pronóstico y tratamiento. El MBCL, homólogo de los linfomas MALT, es una neoplasia recientemente descrita de linfocitos B (302,303,307). |
Estos tumores se distinguen en la mayoría de los casos por una combinación de características morfológicas e inmunofenotípicas específicas (304). El linfoma más frecuente de linfocitos B descrito en pacientes con SS es el MZL, que incluye el linfoma de bajo grado de linfocitos B tipo MALT y el MBCL (249).
La denominación MALT consiste en el tejido linfoide extranodal asociado al epitelio gastrointestinal, bronquial y tejidos de las mucosas. Está compuesto de linfocitos que pueden invadir estos lugares para procesar antígenos luminales y así proporcionar inmunidad de la mucosa. Los linfomas de bajo grado pueden surgir de los tejidos linfoides (MALT-linfomas) y del tejido linfoide asociado a otros tipos de epitelio. Los linfomas MALT se presentan con frecuencia como enfermedad extraganglionar localizada que afecta al tejido epitelial glandular. Isaacson et al (301,305) fueron los primeros en mencionar el concepto de linfomas extranodales MALT. Se ha descrito que este grupo de linfomas surge del tejido extranodal del tubo digestivo, glándulas salivales, pulmón y tiroides (301,305,306) y con poca frecuencia del tejido extranodal [página 111] normal como el de las placas de Peyer. Los linfomas MALT de bajo grado se caracterizan por presentar un curso clínico indolente y una similitud en la organización del MALT normal. Los linfomas MALT poseen características poco conocidas, como el mecanismo por el cual las células neoplásicas permanecen confinadas a un solo lugar, la presencia o ausencia de tráfico neoplásico celular y la diseminación específica de linfomas MALT a otras mucosas. Por otra parte, los linfomas MALT de bajo grado pueden sufrir una transformación histológica a formas más agresivas, habitualmente a un linfoma difuso de células grandes, con el consiguiente cambio en el pronóstico y tratamiento. El MBCL, homólogo de los linfomas MALT, es una neoplasia recientemente descrita de linfocitos B (302,303,307). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [66.] Mpl/Fragment 096 01 - Diskussion Bearbeitet: 7. December 2014, 17:35 Singulus Erstellt: 9. November 2014, 21:32 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 96, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 111, 112, Zeilen: 111: 12 ss.; 112: 1 ss. |
|---|---|
| Las células neoplásicas del MBCL, además de tener inmunoglobulinas monoclonales en la superficie y antígenos B celulares asociados, carecen de CD25 (302,308,309). Clínicamente, el MBCL tiene un curso indolente, afecta a los ganglios linfáticos, puede progresar a un linfoma más agresivo (302,307,309,310) y afectar lugares extranodales (302,309,310). Se sugiere que la asociación frecuente del MBCL con el SS se debe a que las glándulas salivales drenan a los ganglios linfáticos del cuello, que forman parte del ciclo de la circulación sistémica de los linfocitos (311). La asociación de SS y mieloma múltiple (MM) es infrecuente y sólo se han descrito 13 casos (236,244,312-321) (Tabla 16).
TABLA 16. Mieloma múltiple en pacientes con SSp
Entre éstos, dos son plasmocitomas extramedulares de las glándulas salivales (316) y piel (236). También se ha descrito un mieloma parotídeo (314) y un caso de SS como manifestación inicial de un MM (317); la biopsia labial muestra infiltración de linfocitos y células plasmáticas. |
Las células neoplásicas del MBCL, además de tener inmunoglobulinas monoclonales en la superficie y antígenos B celulares asociados, carecen de CD25 (302,308,309). Clínicamente, el MBCL tiene un curso indolente, afecta a los ganglios linfáticos, puede progresar a un linfoma más agresivo (302,307,309,310) y afectar lugares extranodales (302,309,310). Se sugiere que la asociación frecuente del MBCL con el SS se debe a que las glándulas salivales drenan a los ganglios linfáticos del cuello, que forman parte del ciclo de la circulación sistémica de los linfocitos (311).
La asociación de SS y mieloma múltiple (MM) es infrecuente y sólo se han descrito 13 casos (236,244,312-321) (Tabla 16). [página 112] TABLA 16. Mieloma múltiple en pacientes con SSp
Entre éstos, dos son plasmocitomas extramedulares de las glándulas salivales (316) y piel (236). También se ha descrito un mieloma parotídeo (314) y un caso de SS como manifestación inicial de un MM (317); la biopsia labial muestra infiltración de linfocitos y células plasmáticas. |
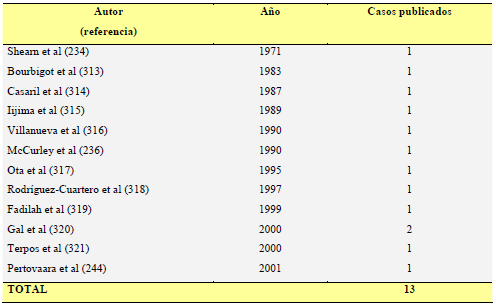
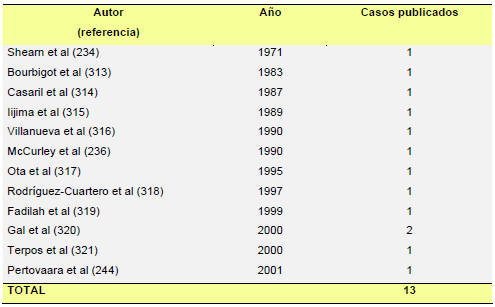
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [67.] Mpl/Fragment 097 01 - Diskussion Bearbeitet: 7. December 2014, 22:29 Singulus Erstellt: 9. November 2014, 21:35 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 97, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 112, 113, Zeilen: 112: 5 ss.; 113: 1 ss. |
|---|---|
| Otras neoplasias hematológicas descritas excepcionalmente en pacientes con SS son 4 casos de MW (132,322,323) y 3 casos de leucemia linfocítica crónica (LLC) (132,323)
b) Linfomas T Los linfomas de linfocitos T se han descrito esporádicamente en pacientes con SS (261,262,324-333) (Tabla 17). TABLA 17. Linfomas T en pacientes con SSp
La presentación más frecuente es la afección cutánea aunque Ros et al (331), han descrito un linfoma T pulmonar angiocéntrico asociado al SS. Finalmente, se ha descrito un caso de linfadenopatía angioinmunoblástica (334). |
Otras neoplasias hematológicas descritas excepcionalmente en pacientes con SS son 4 casos de MW (132,322,323) y 3 casos de leucemia linfocítica crónica (LLC) (132,323)
b) Linfomas T Los linfomas de linfocitos T se han descrito esporádicamente en pacientes con SS (261,262,324-333) (Tabla 17). [página 113] TABLA 17. Linfomas T en pacientes con SSp
La presentación más frecuente es la afección cutánea aunque Ros et al (331), han descrito un linfoma T pulmonar angiocéntrico asociado al SS. Finalmente, se ha descrito un caso de linfadenopatía angioinmunoblástica (334). |
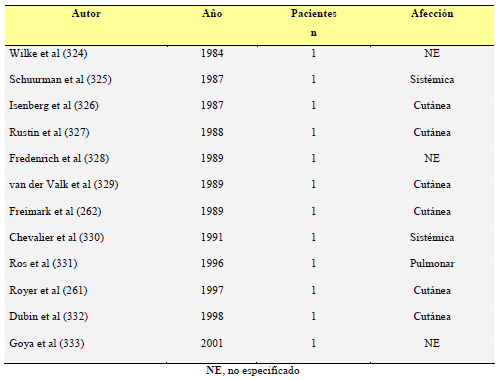
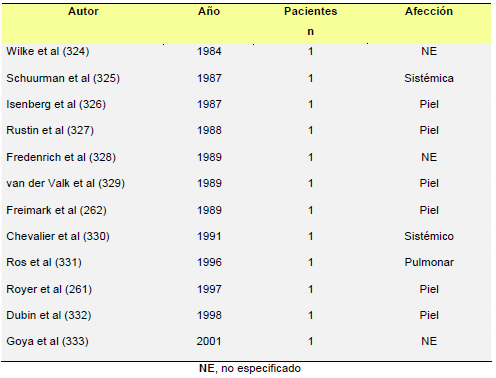
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [68.] Mpl/Fragment 098 01 - Diskussion Bearbeitet: 7. December 2014, 22:33 Singulus Erstellt: 9. November 2014, 21:37 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 98, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 113, 114, Zeilen: 113: 5 ss.; 114: 1 ss. |
|---|---|
| c) Linfoma de Hodgkin
El primer caso de linfoma de Hodgkin (LH) bien documentado en un paciente con SSp se publicó en 1990 (335), aunque esta asociación se había sugerido en estudios previos (228,232,235), no fue hasta este año cuando se describió por completo. En 1992, Vivancos et al (336) describieron dos casos adicionales de LH confirmado por histopatología en pacientes con un diagnóstico clínico e histológico previo de SSp. Estos dos pacientes presentaron diferentes tipos de LH: 1 presentó el tipo de depleción linfocítica y el otro tipo de celularidad mixta. El primer caso publicado (335) fue de predominio linfocítico. Se han reportado otros casos adicionales de SS y LH (11,225,247,335-340) (Tabla 18). TABLA 18. Enfermedad de Hodgkin en pacientes con SSp
Es difícil establecer una relación patogénica entre el SSp y el LH, pues aún se desconoce la causa del LH. |
c) Linfoma de Hodgkin
El primer caso de linfoma de Hodgkin (LH) bien documentado en un paciente con SSp se publicó en 1990 (335), aunque esta asociación se había sugerido en estudios previos (228,232,235), no fue hasta este año cuando se describió por completo. En 1992, Vivancos et al (336) describieron dos casos adicionales de LH confirmado por histopatología en pacientes con un diagnóstico clínico e histológico previo de SSp. Estos dos pacientes [página 114] presentaron diferentes tipos de LH: 1 presentó el tipo de depleción linfocítica y el otro tipo de celularidad mixta. El primer caso publicado (335) fue de predominio linfocítico. Se han reportado otros casos adicionales de SS y LH (11,225,247,335-340) (Tabla 18). TABLA 18. Enfermedad de Hodgkin en pacientes con SSp
Es difícil establecer una relación patogénica entre el SSp y el LH, pues aún se desconoce la causa del LH. |
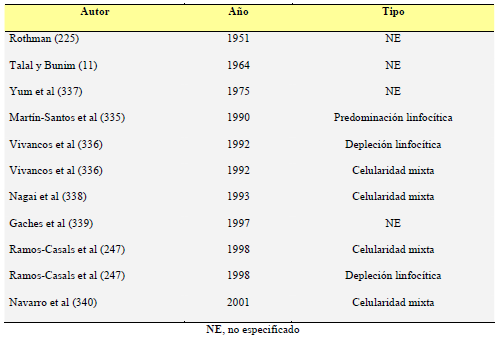
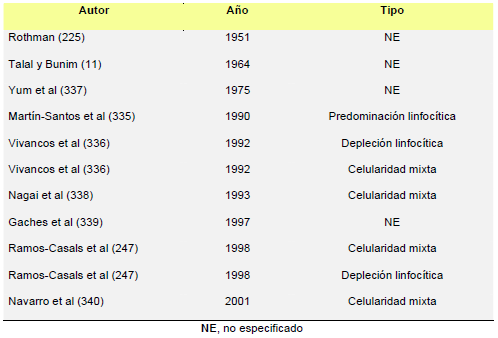
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [69.] Mpl/Fragment 099 01 - Diskussion Bearbeitet: 7. December 2014, 22:35 Singulus Erstellt: 9. November 2014, 21:38 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 99, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 114, 115, Zeilen: 114: 6 ss.; 115: 1 ss. |
|---|---|
| [Estudios recientes han demostrado un origen linfocitario B] para las células de Reed-Sternberg y una predisposición genética en pacientes con SS para el desarrollo de LH similar a la descrita para el linfoma B. Por el contrario, la incidencia de LNH en pacientes con SS es mucho más elevada que la esperada en la población general, a diferencia de la incidencia de LH.
1.7.1.4. Presentación clínica La presentación clínica del linfoma en un paciente con SS es muy diversa. Los LNH con manifestaciones sistémicas se presentan con afección del estado general, hepatoesplenomegalia y linfadenopatía generalizada, lo cual suele estar asociado a estados avanzados de la enfermedad. En un estudio multicéntrico europeo (341), la mayoría de los pacientes con SS y linfoma presentaron parotidomegalia, fiebre, linfadenopatía y una mayor frecuencia de afección extraglandular (vasculitis cutánea y afección del sistema nervioso). De los 33 pacientes, 12 presentaron afección exclusivamente extranodal, 6 afección nodal y 15 ambas afectaciones. Cabe destacar la presencia de adenopatías, sobre todo laterocervicales, en 21 (64%) pacientes. Por otra parte, los focos extranodales más frecuentes fueron las glándulas salivales en 18 pacientes y la gástrica en 4. En la analítica destacaba una elevada frecuencia de citopenias (anemia y leucopenia) y la presencia de inmunoglobulinas monoclonales o crioglobulinas en suero, mientras que la hipogammaglobulinemia fue poco frecuente (menos del 10%). La posible aparición de linfoma en cualquier lugar del organismo donde exista tejido linfoide origina una gran variedad de cuadros clínicos de presentación en el paciente con SS. Los sitios más frecuentes son las glándulas salivales y órganos parenquimatosos como pulmones o tubo digestivo (342) (Tabla 19). |
Estudios recientes han demostrado un origen linfocitario B para las células de Reed-Sternberg y una predisposición genética en pacientes con SS para el desarrollo de LH similar a la descrita para el linfoma B. Por el contrario, la incidencia de LNH en pacientes con SS es mucho más elevada que la esperada en la población general, a diferencia de la incidencia de LH.
[página 115] 2.2.4. Presentación clínica La presentación clínica del linfoma en un paciente con SS es muy diversa. Los LNH con manifestaciones sistémicas se presentan con afección del estado general, hepatoesplenomegalia y linfadenopatía generalizada, lo cual suele estar asociado a estados avanzados de la enfermedad. En un estudio multicéntrico europeo (341), la mayoría de los pacientes con SS y linfoma presentaron parotidomegalia, fiebre, linfadenopatía y una mayor frecuencia de afección extraglandular (vasculitis cutánea y afección del sistema nervioso). De los 33 pacientes, 12 presentaron afección exclusivamente extranodal, 6 afección nodal y 15 ambas afectaciones. Cabe destacar la presencia de adenopatías, sobre todo laterocervicales, en 21 (64%) pacientes. Por otra parte, los focos extranodales más frecuentes fueron las glándulas salivales en 18 pacientes y la gástrica en 4. En la analítica destacaba una elevada frecuencia de citopenias (anemia y leucopenia) y la presencia de inmunoglobulinas monoclonales o crioglobulinas en suero, mientras que la hipogammaglobulinemia fue poco frecuente (menos del 10%). La posible aparición de linfoma en cualquier lugar del organismo donde exista tejido linfoide origina una gran variedad de cuadros clínicos de presentación en el paciente con SS. Los sitios más frecuentes son las glándulas salivales y órganos parenquimatosos como pulmones o tubo digestivo (342) (Tabla 19). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [70.] Mpl/Fragment 100 01 - Diskussion Bearbeitet: 7. December 2014, 22:38 Singulus Erstellt: 9. November 2014, 21:40 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 100, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 116, 117, Zeilen: 116: 1 ss.; 117: 1 ss. |
|---|---|
| En una serie francesa de 16 pacientes con SS y linfoma (261), los sitios más frecuentes de afección fueron la parótida (7 casos), el estómago (3 casos) y el pulmón (3 casos). Se han descrito con menos frecuencia otras presentaciones clínicas como: insuficiencia renal hiperuricémica con hipercalcemia (343), clínica neurológica (habitualmente en linfomas de alto grado), masas renales, testiculares u ováricas y, de forma excepcional, se ha descrito afección del timo (256,309,344-346), la piel (341,347) o los anexos oculares (346,348,349).
TABLA 19. Afección extranodal en pacientes con SSp y LNH. (Ver referencia 342)
1.7.1.5. Diagnóstico a) Evaluación clínica La amplísima diversidad en cuanto a presentación y sitio de afección obliga a una precisa estadificación, mediante evaluación radiológica (TAC o RM) y biopsia de la médula ósea. |
TABLA 19. Afección extranodal en pacientes con SSp y LNH. (Ver referencia 342)
En una serie francesa de 16 pacientes con SS y linfoma (261), los sitios más frecuentes de afección fueron la parótida (7 casos), el estómago (3 casos) y el pulmón (3 casos). Se han descrito con menos frecuencia otras presentaciones clínicas como: insuficiencia renal hiperuricémica con hipercalcemia (343), clínica neurológica (habitualmente en linfomas de alto grado), masas renales, testiculares u ováricas y, de forma excepcional, se ha descrito afección del timo (256,309,344-346), la piel (341,347) o los anexos oculares (346,348,349) (Figura 13). [página 117] [...] 2.2.5. Diagnóstico a) Evaluación clínica La amplísima diversidad en cuanto a presentación y sitio de afección obliga a una precisa estadificación, mediante evaluación radiológica (TAC o RM) y biopsia de la médula ósea. |
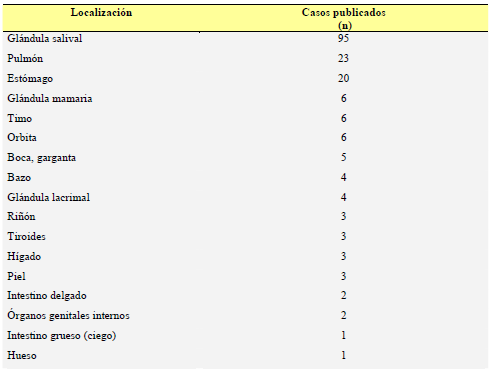

Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [71.] Mpl/Fragment 101 01 - Diskussion Bearbeitet: 7. December 2014, 22:40 Singulus Erstellt: 9. November 2014, 21:43 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 101, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 117, 118, Zeilen: 117: 5 ss.; 118: 1 ss. |
|---|---|
| [La evaluación de datos como la edad, el tamaño de las adenopatías o la] masa linfomatosa, el número de sitios extranodales afectados y los niveles de lactato deshidrogenasa (LDH), contribuyen a determinar el pronóstico y seleccionar la mejor terapéutica individualizada para cada paciente.
El diagnóstico de linfoma, asociado o no a SS, debe fundamentarse siempre con una biopsia tisular, con preferencia de un ganglio linfático. Sólo así será posible obtener el diagnóstico de linfoma y el tipo histológico exacto. La morfología histológica sigue siendo el elemento fundamental para el diagnóstico, pero hoy en día, la inmunohistoquímica y la biología molecular tienen gran importancia en la tipificación de los síndromes linfoproliferativos. El estudio histopatológico continúa siendo básico para el diagnóstico definitivo de linfoma en un paciente con SS. Los datos clínicos pueden resultar poco esclarecedores, ya que la mayoría pueden presentarse en muchos pacientes con SS sin que exista un linfoma. Por otra parte, la existencia de monoclonalidad tampoco puede utilizarse como criterio diagnóstico obligado de malignidad, ya que la expansión clonal es un hecho frecuente en el SS. Por tanto, la sospecha de proliferación linfoide maligna en un paciente con SS obliga a descartar el linfoma mediante el estudio histopatológico. La accesibilidad del tejido varía en función de la sospecha del sitio linfomatoso, que puede ser desde tejidos superficiales (adenopatías periféricas o lesiones cutáneas) hasta órganos parenquimatosos (pulmón o tubo digestivo). Es imprescindible realizar una biopsia a fin de obtener la máxima muestra de tejido. No deben realizarse punciones aspirativas, ya que no son diagnósticas. Distinguir entre un linfoma y una expansión clonal linfocitaria benigna suele ser un problema difícil de resolver, especialmente aquellos casos donde el proceso linfoproliferativo aparece en las mucosas o en las glándulas salivales (301). Además, los linfomas pueden permanecer localizados en un mismo sitio durante muchos años, e [incluso se ha descrito la remisión espontánea sin tratamiento de linfomas de bajo grado (226,227); ello hace todavía más borrosa la línea que separa la benignidad y la malignidad en el linfoma del paciente con SS.] |
La evaluación de datos como la edad, el tamaño de las adenopatías o la masa linfomatosa, el número de sitios extranodales afectados y los niveles de lactato deshidrogenasa (LDH), contribuyen a determinar el pronóstico y seleccionar la mejor terapéutica individualizada para cada paciente.
El diagnóstico de linfoma, asociado o no a SS, debe fundamentarse siempre con una biopsia tisular, con preferencia de un ganglio linfático. Sólo así será posible obtener el diagnóstico de linfoma y el tipo histológico exacto. La morfología histológica sigue siendo el elemento fundamental para el [página 118] diagnóstico, pero hoy en día, la inmunohistoquímica y la biología molecular tienen gran importancia en la tipificación de los síndromes linfoproliferativos. El estudio histopatológico continúa siendo básico para el diagnóstico definitivo de linfoma en un paciente con SS. Los datos clínicos pueden resultar poco esclarecedores, ya que la mayoría pueden presentarse en muchos pacientes con SS sin que exista un linfoma. Por otra parte, la existencia de monoclonalidad tampoco puede utilizarse como criterio diagnóstico obligado de malignidad, ya que la expansión clonal es un hecho frecuente en el SS. Por tanto, la sospecha de proliferación linfoide maligna en un paciente con SS obliga a descartar el linfoma mediante el estudio histopatológico. La accesibilidad del tejido varía en función de la sospecha del sitio linfomatoso, que puede ser desde tejidos superficiales (adenopatías periféricas o lesiones cutáneas) hasta órganos parenquimatosos (pulmón o tubo digestivo). Es imprescindible realizar una biopsia a fin de obtener la máxima muestra de tejido. No deben realizarse punciones aspirativas, ya que no son diagnósticas. Distinguir entre un linfoma y una expansión clonal linfocitaria benigna suele ser un problema difícil de resolver, especialmente aquellos casos donde el proceso linfoproliferativo aparece en las mucosas o en las glándulas salivales (301). Además, los linfomas pueden permanecer localizados en un mismo sitio durante muchos años, e incluso se ha descrito la remisión espontánea sin tratamiento de linfomas de bajo grado (226,227); ello hace todavía más borrosa la línea que separa la benignidad y la malignidad en el linfoma del paciente con SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [72.] Mpl/Fragment 102 01 - Diskussion Bearbeitet: 7. December 2014, 22:42 Singulus Erstellt: 9. November 2014, 21:45 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 102, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 118, 119, Zeilen: 118: 19 ss.; 119: 1 ss. |
|---|---|
| [Además, los linfomas pueden permanecer localizados en un mismo sitio durante muchos años, e] incluso se ha descrito la remisión espontánea sin tratamiento de linfomas de bajo grado (226,227); ello hace todavía más borrosa la línea que separa la benignidad y la malignidad en el linfoma del paciente con SS. Los análisis moleculares, junto al cuadro clínico, pueden ayudar a decidir cuándo un proceso linfoproliferativo benigno como el SS evolucionará hacia la malignidad y cuando precisará de una terapéutica agresiva.
b) Detección de inmunoglobulinas monoclonales circulantes Las inmunoglobulinas monoclonales son el único producto de una sola clona de linfocitos B relativamente maduros o de células plasmáticas. La presencia de inmunoglobulinas monoclonales no equivale por sí misma a la existencia de proliferación linfoide maligna de linfocitos B, sino que indica únicamente la presencia de una o más poblaciones de linfocitos B capaces de producir una inmunoglobulina homogénea. La detección de las inmunoglobulinas monoclonales se realiza por medio de electroforesis de proteínas séricas sobre acetato de celulosa o gel de agarosa que permite en la mayoría de los casos, la detección de una banda electroforéticamente homogénea. Se ha considerado que la característica del proceso de transición de un estado de autoinmunidad a un estado de proliferación linfoide maligno, es la presencia de inmunoglobulinas monoclonales en suero y en orina de pacientes con SS antes de la aparición del linfoma, por lo que su seguimiento y caracterización es de gran valor en pacientes con SS (350,351). En 1983, Moutsopoulus et al (351) encontraron que los pacientes con SS presentaban una elevada incidencia de cadenas ligeras libres circulantes en suero y que el nivel de cadenas ligeras libres en orina se correlacionaba con la actividad de la enfermedad (352). Asimismo, se ha descrito que la expansión monoclonal de linfocitos B en pacientes con SS se correlaciona con la detección de inmunoglobulinas monoclonales o [cadenas ligeras (11,353,354).] |
Además, los linfomas pueden permanecer localizados en un mismo sitio durante muchos años, e incluso se ha descrito la remisión espontánea sin tratamiento de linfomas de bajo grado (226,227); ello hace todavía más borrosa la línea que separa la benignidad y la malignidad en el linfoma del paciente con SS. Los análisis moleculares, junto al cuadro clínico, pueden ayudar a decidir cuándo un proceso linfoproliferativo benigno como el
[página 119] SS evolucionará hacia la malignidad y cuando precisará de una terapéutica agresiva. b) Detección de inmunoglobulinas monoclonales circulantes Las inmunoglobulinas monoclonales son el único producto de una sola clona de linfocitos B relativamente maduros o de células plasmáticas. La presencia de inmunoglobulinas monoclonales no equivale por sí misma a la existencia de proliferación linfoide maligna de linfocitos B, sino que indica únicamente la presencia de una o más poblaciones de linfocitos B capaces de producir una inmunoglobulina homogénea. La detección de las inmunoglobulinas monoclonales se realiza por medio de electroforesis de proteínas séricas sobre acetato de celulosa o gel de agarosa que permite en la mayoría de los casos, la detección de una banda electroforéticamente homogénea. Se ha considerado que la característica del proceso de transición de un estado de autoinmunidad a un estado de proliferación linfoide maligno, es la presencia de inmunoglobulinas monoclonales en suero y en orina de pacientes con SS antes de la aparición del linfoma, por lo que su seguimiento y caracterización es de gran valor en pacientes con SS (350,351). En 1983, Moutsopoulus et al (351) encontraron que los pacientes con SS presentaban una elevada incidencia de cadenas ligeras libres circulantes en suero y que el nivel de cadenas ligeras libres en orina se correlacionaba con la actividad de la enfermedad (352). Asimismo, se ha descrito que la expansión monoclonal de linfocitos B en pacientes con SS se correlaciona con la detección de inmunoglobulinas monoclonales o cadenas ligeras (11,353,354). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [73.] Mpl/Fragment 103 01 - Diskussion Bearbeitet: 7. December 2014, 22:49 Singulus Erstellt: 9. November 2014, 21:47 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 103, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 119, 120, 121, Zeilen: 119: 24; 120: 1 ss.; 121: 1 ss. |
|---|---|
| El origen monoclonal de las células plasmáticas monotípicas en la sialadenitis autoinmune, se ha confirmado mediante estudios sobre el reordenamiento de los genes de las inmunoglobulinas (262,355). Con todo, es necesario mencionar que la evidencia de monoclonalidad no representa necesariamente la existencia de malignidad linfoide. Varios estudios demuestran la presencia de una expansión oligoclonal de linfocitos B en lesiones de las glándulas salivales menores de pacientes con SS sin evolución a linfoma. Pablos et al (356) estudiaron 13 pacientes con SS que presentaban expansión clonal de linfocitos B y ninguno desarrolló enfermedad linfoproliferativa durante el seguimiento. Por tanto, parece que la expansión clonal de los linfocitos B sería sólo un marcador temprano de malignidad y, por lo tanto, no apoya el uso de terapia agresiva en estos pacientes (352,357,358).
Detección del FR monoclonal. Muchos de los pacientes con SS presentan niveles elevados de FR en suero (181). Estudios previos han mostrado que el FR monoclonal presenta idiotipos de reacción cruzada (IRC) (359,360) los cuales se clasifican en 3 grupos: Wa y Po, que incluyen el 60 y 20% del FR monoclonal, respectivamente (359) y el grupo Bla el cual define un subgrupo menor (360). La mayoría de los pacientes con SS expresan un ICR sobre las cadenas κ de sus moléculas de FR definido por el anticuerpo monoclonal 17-109 (258). El Ac 17109 reacciona con el FR monoclonal que pertenece a la familia del idiotipo Wa, y presenta regiones similares variables de la cadena ligera x perteneciente a la cadena del subgrupo VxIIIbx-L y más específico al gen 325 humkv conservado (361) Es importante añadir que la prevalencia de linfocitos B reactivos 17109 dentro de los infiltrados de las glándulas salivales de pacientes con SS es 10 veces mayor que la de ganglios linfáticos de pacientes de la misma edad (258). |
El origen monoclonal de las
[página 120] células plasmáticas monotípicas en la sialadenitis autoinmune, se ha confirmado mediante estudios sobre el reordenamiento de los genes de las inmunoglobulinas (262,355). Con todo, es necesario mencionar que la evidencia de monoclonalidad no representa necesariamente la existencia de malignidad linfoide. Varios estudios demuestran la presencia de una expansión oligoclonal de linfocitos B en lesiones de las glándulas salivales menores de pacientes con SS sin evolución a linfoma. Pablos et al (356) estudiaron 13 pacientes con SS que presentaban expansión clonal de linfocitos B y ninguno desarrolló enfermedad linfoproliferativa durante el seguimiento. Por tanto, parece que la expansión clonal de los linfocitos B sería sólo un marcador temprano de malignidad y, por lo tanto, no apoya el uso de terapia agresiva en estos pacientes (352,357,358). Detección del FR monoclonal. Muchos de los pacientes con SS presentan niveles elevados de FR en suero (181). Estudios previos han mostrado que el FR monoclonal presenta idiotipos de reacción cruzada (IRC) (359,360) los cuales se clasifican en 3 grupos: Wa y Po, que incluyen el 60 y 20% del FR monoclonal, respectivamente (359) y el grupo Bla el cual define un subgrupo menor (360). La mayoría de los pacientes con SS expresan un ICR sobre las cadenas κ de sus moléculas de FR definido por el anticuerpo monoclonal 17-109 (258). El Ac 17109 reacciona con el FR monoclonal que pertenece a la familia del idiotipo Wa, y presenta regiones similares variables de la cadena ligera x perteneciente a la cadena del subgrupo VxIIIbx-L y más específico al gen 325 humkv conservado (361) Es importante añadir que la prevalencia de linfocitos B reactivos 17109 dentro de los infiltrados de las [página 121] glándulas salivales de pacientes con SS es 10 veces mayor que la de ganglios linfáticos de pacientes de la misma edad (258). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [74.] Mpl/Fragment 104 01 - Diskussion Bearbeitet: 7. December 2014, 22:49 Singulus Erstellt: 9. November 2014, 21:50 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 104, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 121, 122, Zeilen: 121: 2 ss.; 122: 1 ss. |
|---|---|
| [Así, los defectos intrínsecos de los linfocitos B] pueden dar como resultado la selección preferencial de un gen que codifique una región variable en particular de la cadena ligera (362) y/o la alteración de diversificar este gen mediante mutaciones somáticas posteriores (363). Las alteraciones de los linfocitos T consisten en la función excesiva de los linfocitos T colaboradores o en la disfunción para expresar clonas particulares de linfocitos B. El mayor número de linfocitos B reactivos frente al MoAb 17-109 en el SS puede formar parte de una alteración generalizada en la inmunoregulación, ya que estos pacientes tienen una alta incidencia de cadenas ligeras libres (351) y paraproteínas en suero y orina (364-366) y en la mayoría de los casos, el FR monoclonal IgM en suero y los crioprecipitados expresan el epítopo asociado con la región variable VkIIIb.
Crioglobulinas monoclonales. Las crioglobulinas mixtas son proteínas que precipitan en frío, están formadas por la IgG y el FR anti-IgG, generalmente una IgM de origen monoclonal (tipo II) o policlonal (tipo III) (367). Las crioglobulinas se han observado en una gran variedad de enfermedades, entre ellas infecciones, enfermedades autoinmunes y neoplasias (368). Desde la publicación del primer caso de crioglobulinemia en un paciente con SS (369), se han realizado algunos estudios sobre la prevalencia y el significado clínico de las crioglobulinas en el SSp. La prevalencia de la crioglobulinemia en el SSp varía desde un 5 a un 61% (144,357,370-374). Las características de los crioprecipitados han demostrado un FR IgMκ monoclonal en la mayoría los pacientes con SS (144,193,371). Ramos et al (144) encontraron que la presencia de vasculitis leucocitoclástica cutánea, la hipocomplementemia y la infección por VHC están independientemente asociados con la presencia de crioglobulinas en el suero de pacientes con SS. En pacientes con SS y crioglobulinemia tipo II, se ha detectado la misma cadena ligera κ en los linfocitos B de las glándulas salivales (370). |
Así, los defectos intrínsecos de los linfocitos B pueden dar como resultado la selección preferencial de un gen que codifique una región variable en particular de la cadena ligera (362) y/o la alteración de diversificar este gen mediante mutaciones somáticas posteriores (363). Las alteraciones de los linfocitos T consisten en la función excesiva de los linfocitos T colaboradores o en la disfunción para expresar clonas particulares de linfocitos B. El mayor número de linfocitos B reactivos frente al MoAb 17-109 en el SS puede formar parte de una alteración generalizada en la inmunoregulación, ya que estos pacientes tienen una alta incidencia de cadenas ligeras libres (351) y paraproteínas en suero y orina (364-366) y en la mayoría de los casos, el FR monoclonal IgM en suero y los crioprecipitados expresan el epítopo asociado con la región variable VkIIIb.
Crioglobulinas monoclonales. Las crioglobulinas mixtas son proteínas que precipitan en frío, están formadas por la IgG y el FR anti-IgG, generalmente una IgM de origen monoclonal (tipo II) o policlonal (tipo III) (367). Las crioglobulinas se han observado en una gran variedad de enfermedades, entre ellas infecciones, enfermedades autoinmunes y neoplasias (368). Desde la publicación del primer caso de crioglobulinemia en un paciente con SS (369), se han realizado algunos estudios sobre la prevalencia y el significado clínico de las crioglobulinas en el SSp. La prevalencia de la crioglobulinemia en el SSp varía desde un 5 a un 61% (144,357,370-374). Las características de los crioprecipitados han demostrado un FR IgMκ monoclonal en la mayoría los pacientes con SS (144,193,371). Ramos et al (144) encontraron que la [página 122] presencia de vasculitis leucocitoclástica cutánea, la hipocomplementemia y la infección por VHC están independientemente asociados con la presencia de crioglobulinas en el suero de pacientes con SS. En pacientes con SS y crioglobulinemia tipo II, se ha detectado la misma cadena ligera κ en los linfocitos B de las glándulas salivales (370). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [75.] Mpl/Fragment 105 01 - Diskussion Bearbeitet: 7. December 2014, 22:51 Singulus Erstellt: 9. November 2014, 21:56 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 105, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 122, 123, Zeilen: 122: 6 ss.; 123: 1 ss. |
|---|---|
| [Moutsopoulus et al (370)] encontraron un aumento de células que expresan las cadenas ligeras κ en los infiltrados de las glándulas salivales menores. Tzioufas et al (375) analizaron la expresión fenotípica de las células plasmáticas que infiltran las glándulas salivales menores de pacientes con SS y encontraron el predominio de células κ positivas, mientras que los pacientes sin crioglobulinas o con crioglobulinas policlonales tenían un porcentaje similar de cadenas κ y λ expresadas en las células plasmáticas (262). Es por ello que la presencia de crioglobulinemia tipo II con un FR IgM monoclonal puede considerarse como un signo temprano de selección oligoclonal/monoclonal de linfocitos B en pacientes con SS.
c) Estudios genéticos y moleculares La evaluación de la expansión clonal de los linfocitos B es un paso preliminar importante para estudiar los eventos patobiológicos implicados en las diferentes etapas de progresión del linfoma. La valoración de la clonalidad depende de la técnica utilizada e incluye estudios de inmunofenotipado e inmunogenotipado para identificar la proliferación monoclonal de los linfocitos B en el tejido linfoide infiltrado (304). Estudios de inmunofenotipado. Las expansiones clonales se caracterizan por reordenamientos de genes idénticos que codifican las inmunoglobulinas o los receptores de linfocitos T, dando como resultado una expresión fenotípica homogénea de esos productos de genes (376). El estudio de inmunofenotipado de los linfocitos periféricos, así como de los linfocitos en los diversos tejidos (ganglios linfáticos, bazo, médula ósea, etc) mediante citometría de flujo, se utiliza ampliamente en el laboratorio para el diagnóstico y la tipificación de las alteraciones linfoproliferativas. La tipificación fenotípica sobre la clonalidad de los linfocitos B implica el uso de anticuerpos [monoclonales o policlonales dirigidos contra las cadenas ligeras y pesadas de las inmunoglobulinas, con la utilización de antisuero específico peroxidasa conjugado para cadenas pesadas (IgM, IgA, IgG) o cadenas ligeras (λ o κ) que se aplican directamente sobre secciones de tejido para identificar poblaciones monoclonales.] |
Moutsopoulus et al (370) encontraron un aumento de células que expresan las cadenas ligeras κ en los infiltrados de las glándulas salivales menores. Tzioufas et al (375) analizaron la expresión fenotípica de las células plasmáticas que infiltran las glándulas salivales menores de pacientes con SS y encontraron el predominio de células κ positivas, mientras que los pacientes sin crioglobulinas o con crioglobulinas policlonales tenían un porcentaje similar de cadenas κ y λ expresadas en las células plasmáticas (262). Es por ello que la presencia de crioglobulinemia tipo II con un FR IgM monoclonal puede considerarse como un signo temprano de selección oligoclonal/monoclonal de linfocitos B en pacientes con SS.
c) Estudios genéticos y moleculares La evaluación de la expansión clonal de los linfocitos B es un paso preliminar importante para estudiar los eventos patobiológicos implicados en las diferentes etapas de progresión del linfoma. La valoración de la clonalidad depende de la técnica utilizada e incluye estudios de inmunofenotipado e inmunogenotipado para identificar la proliferación monoclonal de los linfocitos B en el tejido linfoide infiltrado (304). [página 123] Estudios de inmunofenotipado. Las expansiones clonales se caracterizan por reordenamientos de genes idénticos que codifican las inmunoglobulinas o los receptores de linfocitos T, dando como resultado una expresión fenotípica homogénea de esos productos de genes (376). El estudio de inmunofenotipado de los linfocitos periféricos, así como de los linfocitos en los diversos tejidos (ganglios linfáticos, bazo, médula ósea, etc) mediante citometría de flujo, se utiliza ampliamente en el laboratorio para el diagnóstico y la tipificación de las alteraciones linfoproliferativas. La tipificación fenotípica sobre la clonalidad de los linfocitos B implica el uso de anticuerpos monoclonales o policlonales dirigidos contra las cadenas ligeras y pesadas de las inmunoglobulinas, con la utilización de antisuero específico peroxidasa conjugado para cadenas pesadas (IgM, IgA, IgG) o cadenas ligeras (λ o κ) que se aplican directamente sobre secciones de tejido para identificar poblaciones monoclonales. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [76.] Mpl/Fragment 106 01 - Diskussion Bearbeitet: 7. December 2014, 22:53 Singulus Erstellt: 9. November 2014, 23:24 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 106, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 123, 124, Zeilen: 123: 8 ss.; 124: 1 ss. |
|---|---|
| [La tipificación fenotípica sobre la clonalidad de los linfocitos B implica el uso de anticuerpos] monoclonales o policlonales dirigidos contra las cadenas ligeras y pesadas de las inmunoglobulinas, con la utilización de antisuero específico peroxidasa conjugado para cadenas pesadas (IgM, IgA, IgG) o cadenas ligeras (λ o κ) que se aplican directamente sobre secciones de tejido para identificar poblaciones monoclonales. Se conoce bien la aparición y expresión de las distintas inmunoglobulinas durante la ontogenia de linfocitos B normales y esto permite una clara identificación de las etapas de desarrollo de linfocitos B homólogos neoplásicos (377,378). Así, todas las expansiones de linfocitos B capaces de sintetizar y expresar inmunoglobulinas pueden ser bien tipificadas por su expresión de inmunoglobulina citoplasmática o de membrana, con la característica restricción de la cadena ligera. Además, la intensidad de expresión de la inmunoglobulina de membrana se correlaciona con los niveles de diferenciación de los linfocitos B y obviamente con diferentes tipos de expansión neoplásica de los mismos. De todas formas, las diferencias entre el estudio histopatológico y el inmunofenotipado han llevado a la utilización de los estudios de inmunogenotipado.
Estudios de inmunogenotipado Linfocitos B. El inmunogenotipado es la técnica más sensible para detectar los reordenamientos clonales del DNA en biopsias de tejido, por medio de técnicas de hibridación molecular que utilizan sondas específicas de DNA para las cadenas pesadas y ligeras de las inmunoglobulinas. El DNA se digiere in vitro mediante enzimas de restricción bacterianas, los fragmentos de DNA son separados por electroforesis en gel de agarosa y trasferidos a la membrana donde son expuestos a sondas de DNA específicas para las inmunoglobulinas. La membrana se seca y se realiza la autoradiografía para identificar los reordenamientos de bandas que representan las poblaciones clonales. |
La tipificación fenotípica sobre la clonalidad de los linfocitos B implica el uso de anticuerpos monoclonales o policlonales dirigidos contra las cadenas ligeras y pesadas de las inmunoglobulinas, con la utilización de antisuero específico peroxidasa conjugado para cadenas pesadas (IgM, IgA, IgG) o cadenas ligeras (λ o κ) que se aplican directamente sobre secciones de tejido para identificar poblaciones monoclonales. Se conoce bien la aparición y expresión de las distintas inmunoglobulinas durante la ontogenia de linfocitos B normales y esto permite una clara identificación de las etapas de desarrollo de linfocitos B homólogos neoplásicos (377,378). Así, todas las expansiones de linfocitos B capaces de sintetizar y expresar inmunoglobulinas pueden ser bien tipificadas por su expresión de inmunoglobulina citoplasmática o de membrana, con la característica restricción de la cadena ligera. Además, la intensidad de expresión de la inmunoglobulina de membrana se correlaciona con los niveles de diferenciación de los linfocitos B y obviamente con diferentes tipos de expansión neoplásica de los mismos. De todas formas, las diferencias entre el estudio histopatológico y el inmunofenotipado han llevado a la utilización de los estudios de inmunogenotipado.
[página 124] Estudios de inmunogenotipado Linfocitos B. El inmunogenotipado es la técnica más sensible para detectar los reordenamientos clonales del DNA en biopsias de tejido, por medio de técnicas de hibridación molecular que utilizan sondas específicas de DNA para las cadenas pesadas y ligeras de las inmunoglobulinas. El DNA se digiere in vitro mediante enzimas de restricción bacterianas, los fragmentos de DNA son separados por electroforesis en gel de agarosa y trasferidos a la membrana donde son expuestos a sondas de DNA específicas para las inmunoglobulinas. La membrana se seca y se realiza la autoradiografía para identificar los reordenamientos de bandas que representan las poblaciones clonales. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [77.] Mpl/Fragment 107 01 - Diskussion Bearbeitet: 7. December 2014, 22:57 Singulus Erstellt: 9. November 2014, 23:28 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 107, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 124, 125, 126, Zeilen: 124: 11 ss.; 125: 1 ss.; 126: 5 ss. |
|---|---|
| [Esta técnica puede detectar en el tejido un 1% o más de células] conteniendo el mismo reordenamiento de inmunoglobulina frente al 10% por inmunohistoquímica. El inmunogenotipado incluye el Southern blot, la reacción en cadena de la polimerasa (PCR’) y la hibridación in situ (ISH).
La evaluación molecular de la clonalidad de los linfocitos B por análisis de hibridación Southern blot representó un gran avance en el estudio de la linfomagénesis de linfocitos B. Por medio de esta técnica, se puede demostrar la presencia de poblaciones clonales antes de la aparición del proceso linfoproliferativo (299,355,379,380). Este tipo de estudio requiere una gran cantidad de tejido fresco, lo que limita su utilidad en los pacientes. Fishleder et al (355) demostraron la presencia de reordenamientos de genes de inmunoglobulinas en las lesiones de las glándulas salivales que fueron consideradas como lesiones linfoepiteliales benignas sin afección linfomatosa. Por otra parte, Bodeutsch et al (381) describieron que la evolución a gammapatía sistémica monoclonal o linfoma maligno ocurrió exclusivamente en el subgrupo de pacientes con poblaciones de células plasmáticas monotípicas, con una proporción de κ:λ>3. Recientemente, la PCR’ se ha utilizado con éxito en la valoración de la clonalidad de los linfocitos B en tumores por medio de la amplificación de los reordenamientos de la region VDJ de las cadenas pesadas y cadenas ligeras (313,382,383-385). Linfocitos T. Pocos estudios se han centrado en el estudio del repertorio de linfocitos T (RCT) en pacientes con SS durante el curso de la proliferación linfoide linfocitaria B (386-388). La expansión de los linfocitos B se encuentra mantenida por una estimulación antigénica crónica producida en el microambiente local, como se ha demostrado en el modelo del H.pylori asociado a la proliferación linfoide gástrica de linfocitos B (288,389). |
Esta técnica puede detectar en el tejido un 1% o más de células conteniendo el mismo reordenamiento de inmunoglobulina frente al 10% por inmunohistoquímica. El inmunogenotipado incluye el Southern blot, la reacción en cadena de la polimerasa (PCR) y la hibridación in situ (ISH).
La evaluación molecular de la clonalidad de los linfocitos B por análisis de hibridación Southern blot representó un gran avance en el estudio de la linfomagénesis de linfocitos B. Por medio de esta técnica, se puede demostrar la presencia de poblaciones clonales antes de la aparición del proceso linfoproliferativo (299,355,379,380). Este tipo de estudio requiere una gran cantidad de tejido fresco, lo que limita su utilidad en los pacientes. Fishleder et al (355) demostraron la presencia de reordenamientos de genes de inmunoglobulinas en las lesiones de las glándulas salivales que fueron consideradas como lesiones linfoepiteliales benignas sin afección linfomatosa. Por otra parte, Bodeutsch et al (381) describieron que la evolución a gammapatía sistémica monoclonal o linfoma maligno ocurrió exclusivamente en [página 125] el subgrupo de pacientes con poblaciones de células plasmáticas monotípicas, con una proporción de κ:λ>3. Recientemente, la PCR se ha utilizado con éxito en la valoración de la clonalidad de los linfocitos B en tumores por medio de la amplificación de los reordenamientos de la region VDJ de las cadenas pesadas y cadenas ligeras (313,382,383). [página 126] Linfocitos T. Pocos estudios se han centrado en el estudio del repertorio de linfocitos T (RCT) en pacientes con SS durante el curso de la proliferación linfoide linfocitaria B (386-388). La expansión de los linfocitos B se encuentra mantenida por una estimulación antigénica crónica producida en el microambiente local, como se ha demostrado en el modelo del H.pylori asociado a la proliferación linfoide gástrica de linfocitos B (288,389). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [78.] Mpl/Fragment 108 01 - Diskussion Bearbeitet: 8. December 2014, 12:32 Singulus Erstellt: 9. November 2014, 23:38 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 108, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 126, 127, Zeilen: 126: 10 ss.; 127: 1 ss. |
|---|---|
| [Así, el estudio de la expansión clonal de linfocitos T puede ser] de gran importancia en el estudio de diferentes etapas de la proliferación linfoide de linfocitos B. Para la evaluación de la expresión del RCT, se utiliza la citometría de flujo y/o estudios de inmunoquímica usando anticuerpos monoclonales (390,391).
Se han obtenido resultados diferentes en el estudio de la expansión de los linfocitos T en ciertas enfermedades autoinmunes que predisponen a la malignidad de los linfocitos B como la AR (392) pero pocos estudios se han concentrado en el SS (393,394). Las diferencias metodológicas, la existencia de una gran mayoría de linfocitos T no patológicas en el tejido lesionado, la distinta etapa de la enfermedad, el posible papel de múltiples antígenos ambientales o autoantígenos y la base genética, demuestran el por qué no se ha consensuado la existencia de las funciones del gen RCT (353,391,395,396). 1.7.1.6. Tratamiento No existe una conducta terapéutica única para los pacientes afectos de un linfoma de bajo grado, en la mayoría de casos con linfoma MALT. Si el linfoma afecta a glándulas exocrinas y se encuentra localizado, se ha sugerido la no intervención terapéutica, con vigilancia activa de la enfermedad. Sin embargo, en la mayoría de los casos, se opta por aplicar tratamiento. Las opciones son muy variadas. La cirugía tiene su papel en formas localizadas si el tumor puede extirparse en su totalidad. En estos casos, la cirugía puede ser curativa, si bien el paciente puede desarrollar posteriormente un nuevo linfoma. La radioterapia se usa, en general, como tratamiento complementario si la exéresis quirúrgica no ha podido ser completa o, en ocasiones, tras la quimioterapia. Como excepción, se han de señalar los linfomas parotídeos, en los cuales la radioterapia puede ocasionar mucositis, así como una excacerbación de la xerostomía (397). |
Así, el estudio de la expansión clonal de linfocitos T puede ser de gran importancia en el estudio de diferentes etapas de la proliferación linfoide de linfocitos B. Para la evaluación de la expresión del RCT, se utiliza la citometría de flujo y/o estudios de inmunoquímica usando anticuerpos monoclonales (390,391).
Se han obtenido resultados diferentes en el estudio de la expansión de los linfocitos T en ciertas enfermedades autoinmunes que predisponen a la malignidad de los linfocitos B como la AR (392) pero pocos estudios se han concentrado en el SS (393,394). Las diferencias metodológicas, la existencia de una gran mayoría de linfocitos T no patológicas en el tejido lesionado, la distinta etapa de la enfermedad, el posible papel de múltiples antígenos ambientales o autoantígenos y la base genética, demuestran el por qué no se ha consensuado la existencia de las funciones del gen RCT (353,391,395,396). [página 127] 2.2.6. Tratamiento No existe una conducta terapéutica única para los pacientes afectos de un linfoma de bajo grado, en la mayoría de casos con linfoma MALT. Si el linfoma afecta a glándulas exocrinas y se encuentra localizado, se ha sugerido la no intervención terapéutica, con vigilancia activa de la enfermedad. Sin embargo, en la mayoría de los casos, se opta por aplicar tratamiento. Las opciones son muy variadas. La cirugía tiene su papel en formas localizadas si el tumor puede extirparse en su totalidad. En estos casos, la cirugía puede ser curativa, si bien el paciente puede desarrollar posteriormente un nuevo linfoma. La radioterapia se usa, en general, como tratamiento complementario si la exéresis quirúrgica no ha podido ser completa o, en ocasiones, tras la quimioterapia. Como excepción, se han de señalar los linfomas parotídeos, en los cuales la radioterapia puede ocasionar mucositis, así como una excacerbación de la xerostomía (397). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [79.] Mpl/Fragment 109 01 - Diskussion Bearbeitet: 8. December 2014, 12:46 Singulus Erstellt: 9. November 2014, 23:40 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 109, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 127, 128, Zeilen: 127: 14 ss.; 128: 1 ss. |
|---|---|
| [La quimioterapia es el tratamiento más utilizado, tanto para las formas] diseminadas como para aquellas formas localizadas en las que la cirugía y la radioterapia son muy problemáticas (por ejemplo, los linfomas MALT pulmonares). El tipo de quimioterapia varía según los centros. Desde la monoterapia (clorambucilo o ciclofosfamida, combinados en ocasiones con prednisona), a la poliquimioterapia convencional (de tipo CHOP: ciclofosfamida, adriamicina, vincristina y prednisona). En los últimos años, se han utilizado terapias biológicas, como el α-interferón (por ejemplo, para los linfomas MALT de la conjuntiva ocular) ó actualmente, ciertos anticuerpos monoclonales. El más representativo de estos últimos es el anti-CD20 que ha mostrado su eficacia en los linfomas MALT gástricos y cutáneos.
El tratamiento básico de cualquier linfoma agresivo es, desde luego, la poliquimioterapia. El régimen CHOP es el tratamiento estándar, pero según las circunstancias y los factores pronósticos se pueden administrar quimioterapias más o menos agresivas. En casos seleccionados como en pacientes jóvenes con mal pronóstico, con respuesta parcial o tras una recidiva de un linfoma agresivo, se puede realizar un transplante autólogo con promotores hematopoyéticos. a) Estudio multicéntrico europeo de pacientes con linfoma y SS En la serie multicéntrica europea que estudia las características de 33 pacientes con linfoma y SS (341), los pacientes con linfoma de bajo grado no recibieron tratamiento alguno, y tan sólo uno de ellos progresó a linfoma de alto grado. Los restantes 21 pacientes recibieron tratamiento, 15 con quimioterapia y el resto con radioterapia/cirugía. Se consiguió la remisión completa en 17 (81%), de los cuales 12 eran linfomas de bajo grado, y sólo la remisión parcial en 2. De los 33 pacientes, murieron un total de 9 (27%); 4 (40%) de los 10 pacientes con linfoma de grado intermedio/alto (por progresión de la enfermedad o complicaciones del tratamiento [quimioterapéutico) y 5 (23%) de los 23 pacientes con linfomas de bajo grado (sólo uno progresó a alto grado, el resto murió por causas no relacionadas con el linfoma).] |
La quimioterapia es el tratamiento más utilizado, tanto para las formas diseminadas como para aquellas formas localizadas en las que la cirugía y la radioterapia son muy problemáticas (por ejemplo, los linfomas MALT pulmonares). El tipo de quimioterapia varía según los centros. Desde la monoterapia (dorambucilo [sic] o ciclofosfamida, combinados en ocasiones con prednisona), a la poliquimioterapia convencional (de tipo CHOP: ciclofosfamida, adriamicina, vincristina y prednisona). En los últimos años, se han utilizado terapias biológicas, como el α-interferón (por ejemplo, para los linfomas MALT de la conjuntiva ocular) ó actualmente, ciertos anticuerpos monoclonales. El más representativo de estos últimos es el anti- CD20 que ha mostrado su eficacia en los linfomas MALT gástricos y cutáneos.
[página 128] El tratamiento básico de cualquier linfoma agresivo es, desde luego, la poliquimioterapia. El régimen CHOP es el tratamiento estándar, pero según las circunstancias y los factores pronósticos se pueden administrar quimioterapias más o menos agresivas. En casos seleccionados como en pacientes jóvenes con mal pronóstico, con respuesta parcial o tras una recidiva de un linfoma agresivo, se puede realizar un transplante autólogo con promotores hematopoyéticos. a) Estudio multicéntrico europeo de pacientes con linfoma y SS En la serie multicéntrica europea que estudia las características de 33 pacientes con linfoma y SS (341), los pacientes con linfoma de bajo grado no recibieron tratamiento alguno, y tan sólo uno de ellos progresó a linfoma de alto grado. Los restantes 21 pacientes recibieron tratamiento, 15 con quimioterapia y el resto con radioterapia/cirugía. Se consiguió la remisión completa en 17 (81%), de los cuales 12 eran linfomas de bajo grado, y sólo la remisión parcial en 2. De los 33 pacientes, murieron un total de 9 (27%); 4 (40%) de los 10 pacientes con linfoma de grado intermedio/alto (por progresión de la enfermedad o complicaciones del tratamiento quimioterapéutico) y 5 (23%) de los 23 pacientes con linfomas de bajo grado (sólo uno progresó a alto grado, el resto murió por causas no relacionadas con el linfoma). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [80.] Mpl/Fragment 110 01 - Diskussion Bearbeitet: 8. December 2014, 12:49 Singulus Erstellt: 9. November 2014, 23:44 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 110, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 128, 129, Zeilen: 128: 15 ss.; 129: 1 ss. |
|---|---|
| [De los 33 pacientes, murieron un total de 9 (27%); 4 (40%) de los 10 pacientes con linfoma de grado intermedio/alto (por progresión de la enfermedad o complicaciones del tratamiento] quimioterapéutico) y 5 (23%) de los 23 pacientes con linfomas de bajo grado (sólo uno progresó a alto grado, el resto murió por causas no relacionadas con el linfoma). La supervivencia media fue de 1.8 años para los linfomas de grado intermedio/alto y de 6.33 para los de bajo grado. Finalmente se demostró que la presencia de síntomas B, el tamaño tumoral superior a 7 cm y el tipo histológico, eran factores asociados a una menor supervivencia. En otro estudio, Royer et al (261) consiguieron una remisión completa a los 5 años en 11 de los 16 pacientes, y la remisión parcial de la enfermedad en otros 3.
La localización y el tipo histológico son factores importantes a tener en cuenta en el manejo del linfoma extranodal del paciente con SS, en general, los linfomas gastrointestinales y nasofaríngeos suelen ser más agresivos que los pulmonares, oculares o salivales. La mayoría de los linfomas de glándulas salivales son de bajo grado y son localizados, con una supervivencia que alcanza el 70-80% a los 5 años y del 40-50% a los 10 años. 1.7.2. Mortalidad El riesgo elevado en desarrollar un linfoma en pacientes con SS ha obligado a los investigadores clínicos a intentar establecer factores pronósticos (Tabla 20). |
De los 33 pacientes, murieron un total de 9 (27%); 4 (40%) de los 10 pacientes con linfoma de grado intermedio/alto (por progresión de la enfermedad o complicaciones del tratamiento quimioterapéutico) y 5 (23%) de los 23 pacientes con linfomas de bajo grado (sólo uno progresó a alto grado, el resto murió por causas no relacionadas con el linfoma). La supervivencia media fue de 1.8 años para los linfomas de grado intermedio/alto y de 6.33 para los de bajo grado. Finalmente se demostró que la presencia de síntomas B, el tamaño tumoral superior a 7 cm y el tipo histológico, eran factores asociados a una menor supervivencia. En otro estudio, Royer et al (261) consiguieron una
[página 129] remisión completa a los 5 años en 11 de los 16 pacientes, y la remisión parcial de la enfermedad en otros 3. La localización y el tipo histológico son factores importantes a tener en cuenta en el manejo del linfoma extranodal del paciente con SS, en general, los linfomas gastrointestinales y nasofaríngeos suelen ser más agresivos que los pulmonares, oculares o salivales. La mayoría de los linfomas de glándulas salivales son de bajo grado y son localizados, con una supervivencia que alcanza el 70-80% a los 5 años y del 40-50% a los 10 años. 2.3. Mortalidad El riesgo elevado en desarrollar un linfoma en pacientes con SS ha obligado a los investigadores clínicos a intentar establecer factores pronósticos (Tabla 20). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [81.] Mpl/Fragment 111 01 - Diskussion Bearbeitet: 8. December 2014, 16:15 Singulus Erstellt: 9. November 2014, 23:49 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 111, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 129, 130, Zeilen: 129: tabla; 130: 1 ss. |
|---|---|
| TABLA 20. Características clínicas e inmunológicas que se han relacionado con la aparición de un síndrome linfoproliferativo en el SSp
En 1978, Kassan et al (233) fueron los primeros autores en describir que la existencia de linfadenopatía, esplenomegalia, parotidomegalia y exposición previa a sustancias inmunosupresoras se asociaba al desarrollo de linfoma en pacientes con SS. Han tenido que pasar más de veinte años para que estudios con un número suficiente de pacientes confirmen estos datos. Por ejemplo, en el estudio multicéntrico europeo (341), en el que 33 (27%) pacientes con linfoma y SS habían recibido tratamiento inmunosupresor previo, el 85% presentaban parotidomegalia y el 66% linfadenopatías, aunque sólo el 3% presentaban esplenomegalia. Otro estudio realizado por Valesini et al (242) confirma que la linfadenopatía y la esplenomegalia son factores clínicos predictivos del desarrollo de LNH. Datos parecidos obtuvieron Sutcliffe et al (313), quienes observaron que la historia previa de parotidomegalia y linfadenopatía predecían la evolución a linfoma. Otros estudios han detectado una mayor presencia de manifestaciones extraglandulares en los pacientes que desarrollan linfoma, por ejemplo [púrpura en extremidades inferiores, posiblemente secundaria a la existencia de crioglobulinas (233,239,241,299).] |
TABLA 20. Características clínicas e inmunológicas que se han relacionado con la aparición de un síndrome linfoproliferativo en el SSp
[página 130] En 1978, Kassan et al (233) fueron los primeros autores en describir que la existencia de linfadenopatía, esplenomegalia, parotidomegalia y exposición previa a sustancias inmunosupresoras se asociaba al desarrollo de linfoma en pacientes con SS. Han tenido que pasar más de veinte años para que estudios con un número suficiente de pacientes confirmen estos datos. Por ejemplo, en el estudio multicéntrico europeo (341), en el que 33 (27%) pacientes con linfoma y SS habían recibido tratamiento inmunosupresor previo, el 85% presentaban parotidomegalia y el 66% linfadenopatías, aunque sólo el 3% presentaban esplenomegalia. Otro estudio realizado por Valesini et al (242) confirma que la linfadenopatía y la esplenomegalia son factores clínicos predictivos del desarrollo de LNH. Datos parecidos obtuvieron Sutcliffe et al (313), quienes observaron que la historia previa de parotidomegalia y linfadenopatía predecían la evolución a linfoma. Otros estudios han detectado una mayor presencia de manifestaciones extraglandulares en los pacientes que desarrollan linfoma, por ejemplo púrpura en extremidades inferiores, posiblemente secundaria a la existencia de crioglobulinas (233,239,241,299). |

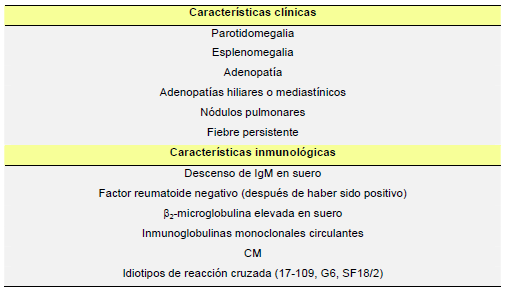
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [82.] Mpl/Fragment 112 01 - Diskussion Bearbeitet: 8. December 2014, 16:19 Singulus Erstellt: 9. November 2014, 23:50 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 112, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 130, 131, Zeilen: 130: 13 ss.; 131: 1 ss. |
|---|---|
| [Otros estudios han detectado una mayor presencia de manifestaciones extraglandulares en los pacientes que desarrollan linfoma, por ejemplo] púrpura en extremidades inferiores, posiblemente secundaria a la existencia de crioglobulinas (233,239,241,299). Finalmente, los pacientes con inicio de la enfermedad antes de los 35 años, presentan una mayor prevalencia de linfadenopatía, FR, crioglobulinas y proliferación linfoide, lo cual podría conferir a la edad de inicio del SS un importante valor pronóstico (247).
En 1971, Cummings et al (398) describieron una reducción importante de la hipergammaglobulinemia justo antes de la aparición del linfoma en pacientes con SS. Otros autores describieron reducciones en los niveles de IgM sérico o negativización del FR (399) datos que no se han confirmado en estudios posteriores. Algunos autores han descrito niveles elevados de β2-microglobulina (147) y del receptor soluble de la IL-2 (400) como marcadores de posible evolución a linfoma. Por último, Tzioufas et al (193) han demostrado recientemente que la presencia de CM y de ciertos idiotipos del FR monoclonal (17-109 y G6) se asocia a un riesgo mayor de desarrollar linfoma. En una serie de 103 pacientes con SS seguidos prospectivamente durante 5 años, 7 desarrollaron linfoma, y 6 de ellos (83%) tenían crioglobulinas al inicio del estudio frente a 12 (12%) de los 96 que al final no desarrollaron linfoma. Estos datos muestran que la determinación de crioglobulinas, un dato de laboratorio fácil de realizar, puede utilizarse como un factor predictivo del posible desarrollo de linfoma en pacientes con SS. |
Otros estudios han detectado una mayor presencia de manifestaciones extraglandulares en los pacientes que desarrollan linfoma, por ejemplo púrpura en extremidades inferiores, posiblemente secundaria a la existencia de crioglobulinas (233,239,241,299). Finalmente, los pacientes con inicio de la enfermedad antes de los 35 años, presentan una mayor prevalencia de linfadenopatía, FR, crioglobulinas y proliferación linfoide, lo cual podría conferir a la edad de inicio del SS un importante valor pronóstico (247).
En 1971, Cummings et al (398) describieron una reducción importante de la hipergammaglobulinemia justo antes de la aparición del linfoma en pacientes con SS. Otros autores describieron reducciones en los niveles de IgM sérico o negativización del FR (399) datos que no se han confirmado en estudios posteriores. Algunos autores han descrito niveles elevados de β2- [página 130] microglobulina (147) y del receptor soluble de la IL-2 (400) como marcadores de posible evolución a linfoma. Por último, Tzioufas et al (193) han demostrado recientemente que la presencia de CM y de ciertos idiotipos del FR monoclonal (17-109 y G6) se asocia a un riesgo mayor de desarrollar linfoma. En una serie de 103 pacientes con SS seguidos prospectivamente durante 5 años, 7 desarrollaron linfoma, y 6 de ellos (83%) tenían crioglobulinas al inicio del estudio frente a 12 (12%) de los 96 que al final no desarrollaron linfoma. Estos datos muestran que la determinación de crioglobulinas, un dato de laboratorio fácil de realizar, puede utilizarse como un factor predictivo del posible desarrollo de linfoma en pacientes con SS. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [83.] Mpl/Fragment 113 01 - Diskussion Bearbeitet: 8. December 2014, 16:25 Singulus Erstellt: 9. November 2014, 23:53 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 113, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 92, 93, Zeilen: 92: 13 ss.; 93: 1 ss. |
|---|---|
| 1.8. Tratamiento
1.8.1. Sequedad de mucosas El tratamiento es sintomático, y se basa en la sustitución de las secreciones ausentes, ya que no parece existir una terapia de fondo que altere el curso evolutivo de la enfermedad. Para la xeroftalmía se deben utilizar lágrimas artificiales 4 a 6 veces al día o instilar colirios que contengan eledoisina o mucolíticos. Es útil utilizar gafas de goma con cámara cerrada para evitar la evaporación de la lágrima durante la noche. Para la xerostomía el paciente puede incrementar la ingesta de agua, o utilizar productos que simulen o estimulen la producción salival. Es aconsejable mantener en la boca, alimentos ácidos no azucarados que incrementen la secreción de saliva (caramelos ácidos, zumo de limón) y evitar los fármacos anticolinérgicos. La higiene ocular y dental es imprescindible para evitar posibles complicaciones (infecciones, caries). En el tratamiento de la xerostomía se puede utilizar saliva artificial y los que mantengan cierta función o reserva glandular, podrán beneficiarse del uso de sialogogos N-Acetilcisteína, Bromexina y Anetholetrithione y de esta forma obtener secreción de sustancias protectoras de la mucosa oral, como enzimas y anticuerpos presentes sólo en la saliva natural (212). Recientemente se ha evaluado la eficacia de dos fármacos antimuscarínicos, la pilocarpina y la cevimelina. La pilocarpina es un agente parasimpático-mimético, con acción agonista de los receptores muscarínicos M3 de las glándulas salivales, con moderada acción beta-adrenérgica (M2), que estimula la secreción glandular exocrina. Para su difusión actual en el tratamiento de la xerostomía en pacientes con SS, han sido claves los estudios recientes de Vivino et al (213) y Papas et al (214), aleatorizados y controlados con placebo en 600 pacientes con SS, que demuestran la eficacia de la pilocarpina en el tratamiento de la xerostomía. |
1.7. Tratamiento (Figura 12)
1.7.1. Sequedad de mucosas El tratamiento es sintomático, y se basa en la sustitución de las secreciones ausentes, ya que no parece existir una terapia de fondo que altere el curso evolutivo de la enfermedad. Para la xeroftalmía se deben utilizar lágrimas artificiales 4 a 6 veces al día o instilar colirios que contengan eledoisina o mucolíticos. Es útil utilizar gafas de goma con cámara cerrada para evitar la evaporación de la lágrima durante la noche. Para la xerostomía el paciente puede incrementar la ingesta de agua, o utilizar productos que simulen o estimulen la producción salival. Es aconsejable mantener en la boca, alimentos ácidos no azucarados que incrementen la secreción de saliva (caramelos ácidos, zumo de [página 93] limón) y evitar los fármacos anticolinérgicos. La higiene ocular y dental es imprescindible para evitar posibles complicaciones (infecciones, caries). En el tratamiento de la xerostomía se puede utilizar saliva artificial y los que mantengan cierta función o reserva glandular, podrán beneficiarse del uso de sialogogos N-Acetilcisteína, Bromexina y Anetholetrithione y de esta forma obtener secreción de sustancias protectoras de la mucosa oral, como enzimas y anticuerpos presentes sólo en la saliva natural (212). Recientemente se ha evaluado la eficacia de dos fármacos antimuscarínicos, la pilocarpina y la cevimelina. La pilocarpina es un agente parasimpático-mimético, con acción agonista de los receptores muscarínicos M3 de las glándulas salivales, con moderada acción beta-adrenérgica (M2), que estimula la secreción glandular exocrina. Para su difusión actual en el tratamiento de la xerostomía en pacientes con SS, han sido claves los estudios recientes de Vivino et al (213) y Papas et al (214), aleatorizados y controlados con placebo en 600 pacientes con SS, que demuestran la eficacia de la pilocarpina en el tratamiento de la xerostomía. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [84.] Mpl/Fragment 114 01 - Diskussion Bearbeitet: 8. December 2014, 16:28 Singulus Erstellt: 9. November 2014, 23:55 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 114, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 93, 94, Zeilen: 93. 16 ss.; 94: 1 ss. |
|---|---|
| [Igualmente nuestro grupo (215) encontró una mejoría de los] síntomas de sequedad, principalmente del área orofaríngea, en el 75% de 100 pacientes con SSp tratados con Salagen® (pilocarpina). La mejoría de los síntomas fue inmediata y proporcional al incremento gradual de la dosis. Aunque la respuesta a la pilocarpina es individual y variable, la dosis utilizada es de 5 mg, 3 a 4 veces al día, siendo necesario en ocasiones, ajustar la dosis para evitar los efectos adversos. Los efectos secundarios están en relación con su acción colinérgica y pueden aparecer en el 10%-30% de los pacientes durante la primera hora tras su administración, siendo los efectos secundarios más frecuentes, la sudoración, los escalofríos o las náuseas, efectos que desaparecen al disminuir la dosis. Ante una complicación o efecto secundario grave, se debe utilizar atropina subcutánea o intravenosa. Debe evitarse en pacientes con asma bronquial activo, iritis, glaucoma de ángulo estrecho y enfermedades cardíacas no controladas. Algunos pacientes, pueden notar también mejoría de la sequedad ocular e incluso cutánea, nasal y vaginal.
La cevimelina es un nuevo agonista muscarínico M3, que en trabajos experimentales ha mostrado una menor afinidad por el receptor muscarínico M2, presente en corazón y pulmón. Se han publicado recientemente dos estudios (216,217) con el mismo diseño (aleatorizados, doble ciego) que comparan diversas dosis de cevimelina con la administración de placebo. La dosis mejor tolerada fue de una cápsula de 30 mg cada 8 horas. Esta dosificación provoca menor sudoración (16-18%) que la pilocarpina utilizada en comprimidos a dosis de 5 mg cada 6 horas, pero una mayor frecuencia de náuseas (16- 21%) o diarrea (14-16%). Las contraindicaciones son las mismas que para la pilocarpina. Para el tratamiento de las complicaciones graves, se debe usar atropina de igual forma. Respecto a la sequedad de otras mucosas, la xerosis cutánea mejora con el uso de cremas hidratantes y se recomienda utilizar protectores labiales. |
Igualmente nuestro grupo (215) encontró una mejoría de los síntomas de sequedad, principalmente del área orofaríngea, en el 75% de 100 pacientes con SSp tratados con Salagen® (pilocarpina). La mejoría de los síntomas fue inmediata y proporcional al incremento gradual de la dosis. Aunque la respuesta a la pilocarpina es individual y variable, la dosis utilizada es de 5 mg, 3 a 4 veces al día, siendo necesario en ocasiones, ajustar la dosis para evitar los efectos adversos. Los efectos secundarios están en relación con su acción colinérgica y pueden aparecer en el 10%-30% de los pacientes durante la primera hora tras su administración, siendo los efectos secundarios más frecuentes, la sudoración, los escalofríos o las náuseas, efectos que desaparecen
[página 94] al disminuir la dosis. Ante una complicación o efecto secundario grave, se debe utilizar atropina subcutánea o intravenosa. Debe evitarse en pacientes con asma bronquial activo, iritis, glaucoma de ángulo estrecho y enfermedades cardíacas no controladas. Algunos pacientes, pueden notar también mejoría de la sequedad ocular e incluso cutánea, nasal y vaginal. La cevimelina es un nuevo agonista muscarínico M3, que en trabajos experimentales ha mostrado una menor afinidad por el receptor muscarínico M2, presente en corazón y pulmón. Se han publicado recientemente dos estudios (216,217) con el mismo diseño (aleatorizados, doble ciego) que comparan diversas dosis de cevimelina con la administración de placebo. La dosis mejor tolerada fue de una cápsula de 30 mg cada 8 horas. Esta dosificación provoca menor sudoración (16-18%) que la pilocarpina utilizada en comprimidos a dosis de 5 mg cada 6 horas, pero una mayor frecuencia de náuseas (16-21%) o diarrea (14-16%). Las contraindicaciones son las mismas que para la pilocarpina. Para el tratamiento de las complicaciones graves, se debe usar atropina de igual forma. Respecto a la sequedad de otras mucosas, la xerosis cutánea mejora con el uso de cremas hidratantes y se recomienda utilizar protectores labiales. |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [85.] Mpl/Fragment 115 01 - Diskussion Bearbeitet: 8. December 2014, 16:31 Singulus Erstellt: 9. November 2014, 23:57 (Hindemith) | Brito Zeron 2006, Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 115, Zeilen: 1 ss. (página entera) |
Quelle: Brito Zeron 2006 Seite(n): 94, 95, Zeilen: 94: 18 ss.; 95: 1 ss. |
|---|---|
| [Respecto a la sequedad] vaginal, debe vigilarse la aparición de infecciones como la candidiasis, tratar la dispareunia con cremas lubricantes y, en mujeres postmenopáusicas, utilizar corticoides tópicos.
1.8.2. Afección extraglandular El arsenal terapéutico que se dispone para el tratamiento de las manifestaciones extraglandulares del SS es muy similar al utilizado en otras enfermedades autoinmunes como el LES, y se basa en la utilización de antiinflamatorios y antipalúdicos para el tratamiento de la afección articular y general, y el uso de corticoides e inmunosupresores para el tratamiento de las manifestaciones viscerales (218). El tratamiento con corticoides o inmunosupresores no ha demostrado ser útil para evitar la progresión del SS. Su empleo estaría solamente justificado en aquellos casos graves de afección sistémica en la que predominan los fenómenos vasculíticos y neurológicos, miopatías inflamatorias y en las fases iniciales de afección renal o pulmonar. En afección grave (GMN proliferativa, alveolitis, polineuropatía progresiva, úlceras o isquemia cutánea) se deben utilizar fármacos inmunosupresores como la ciclofosfamida en "bolus" endovenosos o las gammaglobulinas. La hidroxicloroquina se ha utilizado con buenos resultados en la afección articular de los pacientes con SS. Recientes estudios han analizado otros fármacos. En una pequeña serie de 16 pacientes con SS se ha analizado el efecto del infliximab, aunque el diseño del estudio y el escaso número de pacientes incluidos obligan a una interpretación cautelosa (219). Otros trabajos han analizado el posible papel del interferón (220), la zidovudina (221), la azatioprina (222) o la 2-cloro-2'-deoxiadenosina (223). |
Respecto a la sequedad vaginal, debe vigilarse la aparición de infecciones como la candidiasis, tratar la dispareunia con cremas lubricantes y, en mujeres postmenopáusicas, utilizar corticoides tópicos.
1.7.2. Afección extraglandular El arsenal terapéutico que se dispone para el tratamiento de las manifestaciones extraglandulares del SS es muy similar al utilizado en otras enfermedades autoinmunes como el LES, y se basa en la utilización de antiinflamatorios y antipalúdicos para el tratamiento de la afección articular y [página 95] general, y el uso de corticoides e inmunosupresores para el tratamiento de las manifestaciones viscerales (218). El tratamiento con corticoides o inmunosupresores no ha demostrado ser útil para evitar la progresión del SS. Su empleo estaría solamente justificado en aquellos casos graves de afección sistémica en la que predominan los fenómenos vasculíticos y neurológicos, miopatías inflamatorias y en las fases iniciales de afección renal o pulmonar. En afección grave (GMN proliferativa, alveolitis, polineuropatía progresiva, úlceras o isquemia cutánea) se deben utilizar fármacos inmunosupresores como la ciclofosfamida en "bolus" endovenosos o las gammaglobulinas. La hidroxicloroquina se ha utilizado con buenos resultados en la afección articular de los pacientes con SS. Recientes estudios han analizado otros fármacos. En una pequeña serie de 16 pacientes con SS se ha analizado el efecto del infliximab, aunque el diseño del estudio y el escaso número de pacientes incluidos obligan a una interpretación cautelosa (219). Otros trabajos han analizado el posible papel del interferón (220), la zidovudina (221), la azatioprina (222) o la 2-cloro-2'-deoxiadenosina (223). |
Brito Zerón (2006) no se menciona. Nota que las referencias son iguales en ambos textos (y no se han documentado). |
|
| [86.] Mpl/Fragment 267 22 - Diskussion Bearbeitet: 8. December 2014, 16:39 Singulus Erstellt: 12. November 2014, 20:52 (Hindemith) | Fragment, Gesichtet, Mpl, SMWFragment, Schutzlevel sysop, Sierra 2002, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 267, Zeilen: 22-24 |
Quelle: Sierra 2002 Seite(n): 15, Zeilen: 4 ss. |
|---|---|
| La HTA es el factor de riesgo cardiovascular más importante relacionado con la patología vascular cerebral (435-437), y la coexistencia de otros factores de riesgo cardiovascular (diabetes mellitus, dislipemia,…) aumenta el riesgo de manera [exponencial.]
435. Kannel WB, Wolf PA, Verter MS, McNamara PM. Epidemiologicassessment [sic] of the role of blood pressure in stroke. The Framingham Study. JAMA 1970;214:301-310. 436. MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, et al. Blood pressure, stroke, and coronary heart disease. Part 1, prolonged differences in blood pressure: prospective observational studies corrected for the regressiondilution [sic] bias. Lancet 1990;335:765-774. 437. Stamler J, Stamler R, Neaton JD. Blood pressure, systolic and dyastolic, andcardiovascular [sic] risks. Arch Intern Med 1993;153:598-615. |
La hipertensión arterial (HTA) es el factor de riesgo cardiovascular más importante relacionado con la patología vascular cerebral1-3, y la coexistencia de otros factores de riesgo cardiovascular (diabetes mellitus, dislipemia, tabaquismo, consumo de alcohol) aumenta el riesgo de manera exponencial.
1. Kannel WB, Wolf PA, Verter MS, McNamara PM. Epidemiologic assessment of the role of blood pressure in stroke. The Framingham Study. JAMA 1970;214:301-310. 2. MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, et al. Blood pressure, stroke, and coronary heart disease. Part 1, prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet 1990;335:765-774. 3. Stamler J, Stamler R, Neaton JD. Blood pressure, systolic and dyastolic, and cardiovascular risks. Arch Intern Med 1993;153:598-615. |
Se continua en la próxima página: Mpl/Fragment 268 01 |
|
| [87.] Mpl/Fragment 268 01 - Diskussion Bearbeitet: 8. December 2014, 16:46 Singulus Erstellt: 12. November 2014, 21:11 (Hindemith) | Fragment, Gesichtet, KomplettPlagiat, Mpl, SMWFragment, Schutzlevel sysop, Sierra 2002 |
|
|
|
| Untersuchte Arbeit: Seite: 268, Zeilen: 1-10 |
Quelle: Sierra 2002 Seite(n): 15, 17, Zeilen: 15: 7 ss.; 17: 11 ss. |
|---|---|
| En efecto, es conocido que la HTA multiplica por seis el riesgo de padecer un accidente vascular cerebral (AVC) (438) y, peor aún, más de la tercera parte de los pacientes hipertensos fallece como consecuencia de complicaciones cerebrovasculares, ya sea en relación directa con la elevación de PA, o con la ateromatosis vascular, acelerada y agravada por la HTA.
Las lesiones cerebrovasculares silentes (infartos lacunares, leucoaraiosis) también están asociadas a la presencia de varios de los factores de riesgo vascular (HTA, dislipemia, diabetes mellitus, hábito tabáquico), aunque la HTA es sin duda el más importante (439) Asimismo, la existencia de una enfermedad cardiovascular previa también es un factor de riesgo relacionado con estas lesiones (439). 438. Sierra C, De la Sierra A. Hipertensión arterial: factor de riesgo cardiovascular. Hipertensión 1999; 16: 52-61. 439. Pantoni L, García JH. The significance of cerebral white matter abnormalities 100 years after Binswanger´s report. A review. Stroke 1995;26:1293-1301. |
En efecto, es conocido que la HTA multiplica por seis el riesgo de padecer un accidente vascular cerebral (AVC)4 y, peor aún, más de la tercera parte de los pacientes hipertensos fallece como consecuencia de complicaciones cerebrovasculares, ya sea en relación directa con la elevación de la presión arterial (PA), o con la ateromatosis vascular acelerada y agravada por la HTA.
[página 17] Las lesiones cerebrovasculares silentes (infartos lacunares, leucoaraiosis) también están asociadas a la presencia de varios factores de riesgo vascular (HTA, dislipemia, diabetes mellitus, hábito tabáquico), aunque la HTA es sin duda el más importante8. Asímismo, la existencia de una enfermedad cardiovascular previa también es un factor de riesgo relacionado con estas lesiones8. 4. National Stroke Association. Stroke prevention: the importance of risk factors. Stroke 1991;1:17-20. 8. Pantoni L, García JH. The significance of cerebral white matter abnormalities 100 years after Binswanger´s report. A review. Stroke 1995;26:1293-1301. |
Posiblemente se puede encontrar el texto paralelo también en la publicación Sierra & De la Sierra (1999). Aun cuando fuera así, la referencia 438 no sería suficiente para cubrir todo el texto paralelo. |
|
| [88.] Mpl/Fragment 302 02 - Diskussion Bearbeitet: 8. December 2014, 16:50 Singulus Erstellt: 12. November 2014, 21:16 (Hindemith) | Fragment, Gesichtet, Mpl, SMWFragment, Schutzlevel sysop, Sierra 2002, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 302, Zeilen: 2-6 |
Quelle: Sierra 2002 Seite(n): 24, 26, Zeilen: 24: 20 ss., 26: 1 ss. |
|---|---|
| Se han relacionado tanto con una arteriosclerosis de los vasos perforantes de la sustancia blanca cerebral, siendo la HTA el factor más implicado, así como un posible componente hemodinámico favorecedor de hipoxia (con una disminución de la presión de perfusión) ocasionado por la pérdida de la autorregulación cerebral observada en la HTA. | Por tanto, las LSB representan fenómenos isquémicos relacionados tanto con una arteriosclerosis de los vasos perforantes de la sustancia blanca cerebral, siendo la HTA el factor más implicado, así como un posible componente hemodinámico favorecedor de hipoxia (con una disminución de la
[página 26] presión de perfusión) ocasionado por la pérdida de la autorregulación cerebral observada en la HTA. |
No se menciona la fuente. |
|