Angaben zur Quelle [Bearbeiten]
| Autor | Sybille Susanne Regn |
| Titel | Simultane Expansion zytotoxischer T-Lymphozyten gerichtet gegen Adeno-und Epstein-Barr-Virus Epitope, zur Therapie von opportunistischen Infektionen nach allogener hämatopoetischer Stammzelltransplantation (HSCT). |
| Jahr | 2002 |
| Anmerkung | Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. |
| URL | https://mediatum.ub.tum.de/doc/601242/601242.pdf |
Literaturverz. |
nein |
| Fußnoten | ja |
| Fragmente | 30 |
| [1.] Dml/Fragment 015 13 - Diskussion Zuletzt bearbeitet: 2018-03-10 22:53:39 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 15, Zeilen: 13-32 |
Quelle: Regn 2002 Seite(n): 14, 15, Zeilen: 14: 25 ff.; 15: 1 ff. |
|---|---|
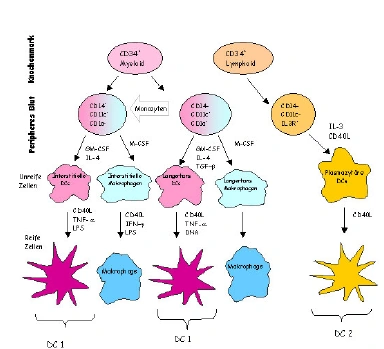
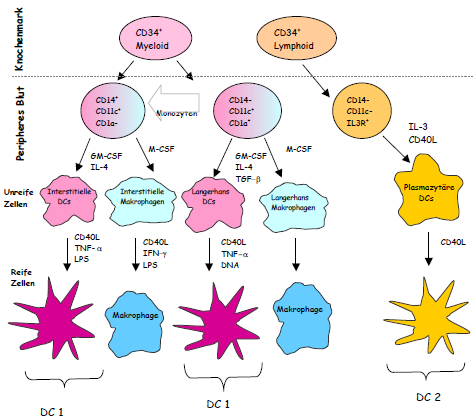
| Obwohl alle Zellen des Körpers die Kapazität aufweisen, Antigene auf ihren Oberflächen zu exprimieren, ist nur eine gewisse Gruppe von hämatopoetischen Zellen in der Lage [sic] als „professionelle“ APCs zu fungieren, um effektiv T-Lymphozyten zu aktivieren und eine Immunantwort zu induzieren. Zu diesen Zelltypen gehören Makrophagen, aktivierte B-Lymphozyten und die dendritischen Zellen (DC), die sich in Langerhans´sche Zellen, interstitielle DCs und lymphoide bzw. plasmazytäre DCs unterteilen lassen. Die dendritischen Zellen sind die potentesten APCs [sic] die bekannt sind [sic] und entwickeln sich ebenfalls aus CD34+ myeloischen Vorläuferzellen des Knochenmarks. Im peripheren Blut beträgt der Anteil an DCs ca. 0,1 –0,5 % aller Leukozyten (Fearnley et al. 1997) [sic] und deshalb wurde die Rolle der DCs in immunologischen Prozessen erst relativ spät erkannt. Nach ihrer Migration aus dem Knochenmark differenzieren sie sich im peripheren Blut zu unreifen DCs und wandern in die angrenzenden Organe, wo sie Antigene aufnehmen. Im Zuge der Antigenaufnahme erfolgt die Aktivierung und Reifung der DCs und folglich die Migration durch die afferenten Lymphgefäße in die T-Zell reichen Regionen der sekundären lymphatischen Organe. Diese Prozesse werden im wesentlichen durch das Zusammenspiel von Chemokinen und Zytokinen und deren Rezeptoren auf den DCs gesteuert. Aufgrund des Fehlens von spezifischen Oberflächenmarkern, die eindeutig unreife DC Vorläuferzellen charakterisieren, ist die Unterscheidung der beiden myeloischen Subpopulationen [noch immer nicht komplett definiert.] | Obwohl alle Zellen des Körpers die Kapazität aufweisen Antigene auf ihren Oberflächen zu exprimieren, ist nur eine gewisse Gruppe von hämatopoetischen Zellen in der Lage [sic] als „professionelle“ APCs zu fungieren, um effektiv T-Lymphozyten zu aktivieren und eine Immunantwort zu induzieren. Zu diesen Zelltypen gehören Makrophagen, aktivierte B-Lymphozyten und die dendritischen Zellen (DC), die sich in Langerhans´sche Zellen, interstitielle DCs und lymphoide bzw. plasmazytäre DCs unterteilen lassen. Die dendritischen Zellen sind die potentesten APCs [sic] die man kennt [sic] und entwickeln sich ebenfalls aus CD34+ myeloischen Vorläuferzellen des Knochenmarks (Abbildung 2). Im peripheren Blut beträgt der Anteil an DCs ca. 0,1 –0,5 % aller Leukozyten (Fearnley et al. 1997) [sic] und deshalb wurde die Rolle der DCs in immunologischen Prozessen erst relativ spät erkannt. Nach ihrer
[Seite 15] Migration aus dem Knochenmark differenzieren sie sich im peripheren Blut zu unreifen DCs und wandern in die angrenzenden Organe, wo sie Antigene aufnehmen. Im Zuge der Antigenaufnahme erfolgt die Aktivierung und Reifung der DCs und folglich die Migration durch die afferenten Lymphgefäße in die T-Zell reichen Regionen der sekundären lymphatischen Organe (Banchereau et al. 1998). Diese Prozesse werden im wesentlichen durch das Zusammenspiel von Chemokinen und Zytokinen und deren Rezeptoren auf den DCs gesteuert. Aufgrund des Fehlens von spezifischen Oberflächenmarkern, die eindeutig unreife DC Vorläuferzellen charakterisieren, ist die Unterscheidung der beiden myeloischen Subpopulationen noch immer nicht komplett definiert. |
Ein Verweis auf die Quelle fehlt. |
|
| [2.] Dml/Fragment 016 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 00:50:17 Hindemith | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 16, Zeilen: 1ff (komplett) |
Quelle: Regn 2002 Seite(n): 15, 16, Zeilen: 15: 9ff; 16: Abbildung |
|---|---|
| Ein Unterschied besteht darin, daß sie in unterschiedlichen Geweben des Körpers vorkommen. Die Langerhans´schen Zellen befinden sich vornehmlich in der Haut, während die interstitiellen DCs in allen Organen vorkommen, um dort Antigene aufzunehmen. Beide Populationen dirigieren CD-4+ T-Lymphozyten vornehmlich in Richtung sogenannter T-Helfer 1 (TH1)- Antworten (Arpinati et al. 2000, Cella et al. 1996). Die dritte Population, die lymphoiden oder plasmazytären ( CD123+ und CD8+) DCs kommen im Blut und in den Tonsillen vor und zeigen eine starke Abhängigkeit von dem Zytokin Interleukin-3 (Banchereau et al. 2000, Galibert et al. 2001). Sie dirigieren CD-4+ T-Zellen in Richtung T-Helfer 2 (TH2)- Aktivität und werden daher auch als DC2 (Arpinati et al. 2000) bezeichnet. Damit ist ihre Rolle stark mit dem humoralen Arm der adaptiven Immunität verknüpft.
Abbildung 2. Schematische Darstellung der Differenzierung verschiedener Subpopulationen von dendritischen Zellen, aus einer CD34+ Vorläuferzelle, in Abhängigkeit des umgebenden Zytokin-Mikro-Milieus (nach Banchereau et al. 2000). |
Ein Unterschied besteht darin, daß sie in unterschiedlichen Geweben des Körpers vorkommen. Die Langerhans´schen Zellen befinden sich vornehmlich in der Haut, während die interstitiellen DCs in allen Organen vorkommen um dort Antigene aufzunehmen. Beide Populationen dirigieren CD4+ T-Lymphozyten vornehmlich in Richtung sogenannter T-Helfer 1 (TH1)- Antworten (Arpinati et al. 2000, Cella et al. 1996). Die dritte Population, die lymphoiden oder plasmazytären (CD123+ und CD8+) DCs kommen im Blut und in den Tonsillen vor und zeigen eine starke Abhängigkeit von dem Zytokin Interleukin-3 (Banchereau et al. 2000, Galibert et al. 2001). Sie dirigieren CD4+ T-Zellen in Richtung T-Helfer 2 (TH2)- Aktivität und werden daher auch als DC2 (Kuwana et al. 2001, Arpinati et al. 2000) bezeichnet. Damit ist ihre Rolle stark mit dem humoralen Arm der adaptiven Immunität verknüpft.
[Seite 16]
Abbildung 2: Schematische Darstellung der Differenzierung verschiedener Subpopulationen von dendritischen Zellen, aus einer CD34+ Vorläuferzelle, in Abhängigkeit des umgebenden Zytokin-Mikro-Milieus |
Ein Verweis auf die Quelle fehlt. |
|
| [3.] Dml/Fragment 017 01 - Diskussion Zuletzt bearbeitet: 2018-03-12 17:28:52 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 17, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 17, Zeilen: 1 ff. |
|---|---|
| DCs spielen eine zentrale Rolle bei der Antigenpräsentation. Hochaffine antigenbindende Rezeptoren, wie der Mannoserezeptor und die Fcγ und Fcε- Rezeptoren erlauben eine Antigenaufnahme von hoher Effizienz, so daß es der Zelle gestattet, Antigen sogar im pico- sowie nanomolaren Bereich zu präsentieren, während andere APCs erst bei einer Antigenkonzentration im mikromolaren Bereich eine effiziente Antigenpräsentation leisten können.
DCs sind die einzigen APCs, die in der Lage sind primäre Immunantworten hervorzurufen und sogar naive CD-8+ zytotoxische T-Zellen zu stimulieren (Bell et al 1999, Banchereau et al. 1998). In vitro Studien haben deutlich demonstrieren können, daß DCs in der Lage waren hoch effizient antigenspezifische CTL-Antworten hervorzurufen. Hierzu wurden sie entweder (i,) genetisch modifiziert (Brossart et al. 1997, Song et al. 1997), (ii,) mit Peptiden oder mit Tumorantigenen (Ashley et al. 1997, Boczkowski et al. 1998, Nair et al. 1998) beladen oder (iii,) mit tumorspezifischer RNA (Nair et al. 1998, Su et al. 2001) beladen um spezifische Antigene zu exprimieren. Experimentelle Ergebnisse von Studien an Tiermodellen (Zitvogel et al. 1996), sowie erste klinische Studien (Nestle et al. 1998, Murphy et al. 1996, Tjoa et al. 1998, Hsu et al. 1996) unterstützen die Hypothese, das [sic] Individuen, die mit Tumorpeptid-beladenen DCs vakziniert wurden, eine tumorspezifische humorale und zelluläre Antwort hervorrufen konnten. Dies führte zum Rückgang der Tumorlast und dem Aufbau einer schützenden Immunität gegenüber neuem Tumorwachstum in vivo. DCs zirkulieren im peripheren Blut und sind aufgrund ihrer hohen Mobilität in sämtlichen Geweben des Körpers zu finden. Da sie in nur geringen Mengen im peripheren Blut vorhanden sind, kann eine direkte Manipulation dieser Zellen in vitro nicht vorgenommen werden, doch haben sich mittlerweile Methoden etabliert, die ein relativ einfaches Anreichern dieser Zellen aus dem peripheren Blut ermöglicht. Durch die Kombination von GM-CSF und Interleukin-4 (IL-4) lassen sich ausreichende Mengen an unreifen DCs aus monozytären Vorläuferzellen des peripheren Blutes differenzieren, deren weitere Entwicklung zu reifen DCs durch Kombination mit TNF− α, GM-CSF und IL-1β erreicht werden kann. Morphologisch sind DCs charakterisiert als bizarr aussehende, unregelmäßig geformte Zellen mit großen Zellkörpern und langen zytoplasmatischen Ausläufern (Dendriten), die sich wie feine Schleierfäden vom [Zellkörper abheben.] |
DCs spielen eine zentrale Rolle bei der Antigenpräsentation. Hochaffine antigenbindende Rezeptoren, wie den [sic] Mannoserezeptor und die Fcγ und Fcε-Rezeptoren erlauben eine Antigenaufnahme von derartiger Effizienz, daß es der Zelle gestattet Antigen sogar im pico oder nanomolaren Bereich zu präsentieren, während andere APCs erst bei einer Antigenkonzentration im mikromolaren Bereich eine effiziente Antigenpräsentation leisten können.
DCs sind die einzigen APCs, die in der Lage sind primäre Immunantworten hervorzurufen und sogar naive CD8+ zytotoxische T-Zellen zu stimulieren (Bell et al 1999, Banchereau et al. 1998). In vitro Studien haben deutlich demonstrieren können, daß DCs, die entweder genetisch modifiziert wurden um spezifische Antigene zu exprimieren (Brossart et al. 1997, Song et al. 1997), mit Peptiden oder mit Tumorantigenen (Ashley et al. 1997, Boczkowski et al. 1998, Nair et al. 1998) beladen oder mit tumorspezifischer RNA (Nair et al. 1998, Su et al. 2001) gepulsed wurden, in der Lage waren hoch effizient antigenspezifische ZTL-Antworten hervorzurufen. Experimentelle Ergebnisse von Studien an Tiermodellen (Zitvogel et al. 1996), sowie erste klinische Studien (Nestlé et al. 1998, Murphy et al. 1996, Tjoa et al. 1998, Hsu et al. 1996) unterstützen die Hypothese, daß Individuen, die mit Tumorpeptid-beladenen DCs vacciniert wurden, eine tumorspezifische humorale und zelluläre Antwort hervorrufen konnten. Dies führte zum Rückgang der Tumorlast und dem Aufbau einer schützenden Immunität gegenüber neuem Tumorwachstum in vivo. DCs zirkulieren im peripheren Blut und sind aufgrund ihrer hohen Mobilität in sämtlichen Geweben des Körpers zu finden. Da sie in nur geringen Mengen im peripheren Blut vorhanden sind, kann eine direkte Manipulation dieser Zellen in vitro nicht vorgenommen werden, doch haben sich mittlerweile Methoden etabliert, die ein relativ einfaches Anreichern dieser Zellen aus dem peripheren Blut ermöglicht. Durch die Kombination von GM-CSF und Interleukin-4 (IL-4) lassen sich ausreichende Mengen an unreifen DCs aus monozytären Vorläuferzellen des peripheren Blutes differenzieren, deren weitere Entwicklung zu reifen DCs durch Kombination mit TNF [sic], GMCSF und IL-1β erreicht werden kann. Morphologisch sind DCs charakterisiert als bizarr aussehende, unregelmäßig geformte Zellen mit großen Zellkörpern und langen zytoplasmatischen Ausläufern (Dendriten), die sich wie feine Schleierfäden vom Zellkörper abheben. |
Ein Verweis auf die Quelle fehlt. |
|
| [4.] Dml/Fragment 018 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 22:44:25 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 18, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 17, 18, Zeilen: 17: letzte vier Zeilen; 18: 1 ff. |
|---|---|
| Im unreifen Zustand zeigen DCs deutlich verkürzte Fortsätze (Dendriten), doch im ausgereiften Stadium werden die zytoplasmatischen Ausläufer länger und nehmen dadurch die eher charakteristische Struktur an.
Die Antigenaufnahme erfolgt normalerweise in der Peripherie und veranlasst dann die DCs zu den sekundären lymphoiden Organen zu wandern, wo sie das Antigen den T-Zellen präsentieren. Folgende Mechanismen der Antigenaufnahme sind charakteristisch für DCs:
|
Im unreifen Zustand zeigen DCs deutlich verkürzte Fortsätze (Dendriten), doch im ausgereiften Stadium werden die zytoplasmatischen Ausläufer länger und nehmen dadurch die eher charakteristische Struktur an.
[Seite 18] Die Antigenaufnahme erfolgt normalerweise in der Peripherie und veranlaßt dann die DCs zu den sekundären lymphoiden Organen zu wandern, wo sie das Antigen den T-Zellen präsentieren. Folgende Mechanismen der Antigenaufnahme sind charakteristisch für DCs: 1. Makropinozytose, 2. Rezeptor vermittelnde Endozytose über den Mannoserezeptor: Der Mannoserezeptor entläßt seinen Ligand bei saurem PH in endosomale Kompartimente und wird dann wieder zurück gewonnen. 3. Rezeptor vermittelnde Endozytose über FcRII- Rezeptoren. Damit ist die DC u.a. in der Lage Antikörper-Antigenkomplexe zu binden und aufzunehmen. 4. Phagozytotische Aufnahme von apoptotischen (Albert et al. 1998) und nekrotischen Zellfragmenten (Galucci et al. 1999), sowie ganzen Zellen über CD36 und αvβ3 und v5 [sic] Integrine. 5. Aufnahme von Viren, Bakterien (auch Mykobakterien) und intrazelluläre Parasiten, wie Leishmania major. 6. Aufnahme von Peptidgeladenen Hitzeschockproteine gp96 und Hsp 70 (Bell 1998). |
Ein Verweis auf die Quelle fehlt. |
|
| [5.] Dml/Fragment 019 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 18:36:10 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 19, Zeilen: 1-4, 12-17 |
Quelle: Regn 2002 Seite(n): 18, 25, Zeilen: 18: 17 ff.; 25: Abb. 6 |
|---|---|
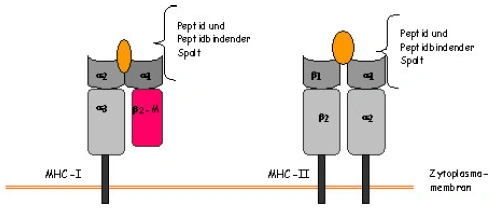
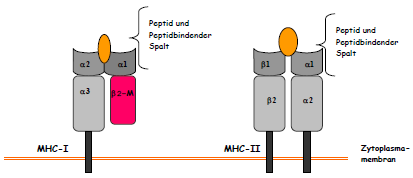
Abbildung 3. MHC-Klasse I-Moleküle bestehen aus zwei Polypeptidketten, eine α- oder schwere Kette, die im MHC-kodiert wird und eine kleinere, nicht kovalent angelagerte Kette, (β2-Mikroglobulin (β2-M)), die nicht im MHC kodiert wird. Nur die α-Kette ist fest in der Zellmembran verankert und besteht aus drei Domänen. Die Struktur der α1 und α2 Domänen bilden ein Paar und schaffen an der Oberfläche des Moleküls den Peptidbindenden Spalt. MHC-Klasse II- Moleküle bestehen aus einem nichtkovalenten Komplex zweier α und β –Ketten, die beide die Membran durchdringen. Beide Ketten haben jeweils zwei Domänen, die gemeinsam den peptidbindenden Spalt bilden (nach Janeway 1997, verändert) Wenn das antigene Protein in den endozytotischen Apparat der Zelle eintritt, wird es bei Makrophagen zunächst in lysosomale Vesikel geschleust, wo es in Peptidfragmente zerlegt wird, die daraufhin auf wenige MHC-Klasse-II-Moleküle geladen werden (Banchereau und Steinman 1998). [...] Weiterhin werden bei DCs große Mengen an MHC-Klasse-II-Moleküle produziert, die in zahlreichen spezialisierten MHC-Klasse-II-reichen Kompartimenten, den MIIC-Vesikeln auf ihre Beladung mit Peptidantigenen warten. Sie kommen nur in unreifen DCs vor und stellen späte endosomale Strukturen dar, die zusätzlich über die notwendige Ausrüstung an HLA-DM-Molekülen verfügen, die dabei helfen das Peptid auf die Bindungsfurche der MHC-II Moleküle zu laden. |
Wenn das antigene Protein in den endozytotischen Apparat der Zelle eintritt, wird es bei Makrophagen zunächst in lysosomale Vesikel geschleust, wo es in Peptidfragmente zerlegt wird, die daraufhin auf wenige MHC-Klasse-II-Moleküle geladen werden (Banchereau und Steinman 1998). Bei DCs dagegen werden riesige Mengen an MHC-Klasse-II-Moleküle produziert, die in zahlreichen spezialisierten MHC-Klasse-II-reichen Kompartimenten, den MIIC-Vesikeln auf ihre Beladung mit Peptidantigenen warten. Sie kommen nur in unreifen DCs vor und stellen späte endosomale Strukturen dar, die zusätzlich über die notwendige Ausrüstung an HLA-DM-Molekülen verfügen, die dabei helfen das Peptid auf die Bindungsfurche der MHC-II Moleküle zu laden.
[Seite 25]
Abbildung 6: MHC-Klasse I-Moleküle bestehen aus zwei Polypeptidketten, eine α- oder schwere Kette, die im MHC-kodiert wird und eine kleinere, nicht kovalent angelagerte Kette, (β2-Mikroglobulin (β2-M)), die nicht im MHC kodiert wird. Nur die α-Kette ist fest in der Zellmembran verankert und besteht aus drei Domänen. Die Struktur der α1 und α2 Domänen bilden ein Paar und schaffen an der Oberfläche des Moleküls den Peptidbindenden Spalt. MHC-Klasse II- Moleküle bestehen aus einem nichtkovalenten Komplex zweier α und β –Ketten, die beide die Membran durchdringen. Beide Ketten haben jeweils zwei Domänen, die gemeinsam den peptidbindenden Spalt bilden (nach Janeway 1997, verändert) |
Ein Verweis auf die Quelle fehlt. |
|
| [6.] Dml/Fragment 020 01 - Diskussion Zuletzt bearbeitet: 2018-03-12 17:37:41 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 20, Zeilen: 1-23 |
Quelle: Regn 2002 Seite(n): 18, 19, Zeilen: 18: 25 ff.; 19: 1 ff. |
|---|---|
| [Der] Antigenaufnahme folgt dann die Reife der DCs und die MIIC-Vesikel entlassen ihre Peptidbeladenen MHC-II-Komplexe an die Zelloberfläche. Diese Oberflächenkomplexe bleiben über mehrere Tage stabil. Darüber hinaus kommt es während der Reifung zur Hochregulierung der für die T-Zellaktivierung entscheidenden Adhäsions- und kostimulatorischen Molekülen, wie CD-54 (ICAM-1), CD-11a (LFA-1), CD-11c, CD-86 (B7-2) und CD-40, sowie zur Produktion von Interleukin-12 (IL-12). Dieses Zytokin hat eine positive immunstimulatorische Funktion und wirkt sowohl auf NK-Zellen, als auch auf B- und T-Zellen. So ausgerüstet erfüllen DCs alle Voraussetzungen um effektiv CD-4+ T-Zellen zu stimulieren. Sie sind allerdings auch in der Lage direkt zytotoxische CD-8+ T-Zellen zu stimulieren, die unmittelbar infizierte Zellen, Tumorzellen oder allogene Transplantate angreifen können. Dabei müssen DCs das Antigen über einen bisher ungeklärten Vorgang an den endosomalen Vesikeln vorbeischleusen, um es über das Proteasom zu prozessieren und über die TAP-Transporter- MHC-I-Exportroute den CD-8+ T-Zellen zu präsentieren. Dabei können sie auch selbst von dem Virus infiziert werden (z.B. Influenza Virus) oder sie sind in der Lage Peptide von nicht replizierenden Mikroben oder sterbenden infizierten Zellen aufzunehmen und die Antigene über den MHC-I-Weg den CD-8+ T-Zellen präsentieren (Banchereau und Steinman 1998, Albert et al. 1998). Aufgrund der hohen Dichte an sowohl MHC-I, als auch MHC-II Molekülen und der Bereitstellung kostimulatorischer Signale ist eine einzige reife DC in der Lage 100 bis 3000 T-Zellen zu aktivieren (Banchereau und Steinman 1998, Bell et al. 1999). | Der Antigenaufnahme folgt dann die Reife der DCs und die MIIC-Vesikel entlassen ihre Peptidbeladenen MHC-II-Komplexe an die Zelloberfläche. Diese Oberflächenkomplexe bleiben über mehrere Tage stabil. Darüber hinaus kommt es während der Reifung zur Hochregulierung der für die T-Zellaktivierung entscheidenden Adhäsions- und Kostimulatorischen Molekülen, wie CD54 (ICAM-1), CD11a (LFA-1), CD11c, CD86 (B7-2) und CD40, sowie zur Produktion von Interleukin-12 (IL-12). Dieses Zytokin hat eine positive immunstimulatorische Funktion und wirkt sowohl auf NK-Zellen, als auch auf B- und T-Zellen. So ausgerüstet erfüllen DCs alle Voraussetzungen um effektiv CD4+ T-Zellen zu stimulieren. Sie sind allerdings auch in der Lage direkt zytotoxische CD8+ T-Zellen zu stimulieren, die unmittelbar infizierte Zellen, Tumorzellen
[Seite 19] oder allogene Transplantate angreifen können. Dabei müssen DCs das Antigen über einen bisher ungeklärten Vorgang an den endosomalen Vesikeln vorbeischleusen, um es über das Proteasom zu prozessieren und über die TAP-Transporter- MHC-I-Exportroute den CD8+ T-Zellen zu präsentieren. Dabei können sie auch selbst von dem Virus infiziert werden (z.B. Influenza Virus) oder sie sind in der Lage Peptide von nicht replizierenden Mikroben oder sterbenden infizierten Zellen aufzunehmen und die Antigene über den MHC-I-Weg den CD8+ T-Zellen präsentieren (Banchereau und Steinman 1998, Albert et al. 1998). Aufgrund der hohen Dichte an sowohl MHC-I, als auch MHC-II Molekülen und der Bereitstellung kostimulatorischer Signale ist ein [sic] einzige reife DC in der Lage 100 bis 3000 T-Zellen zu aktivieren. Dies macht sie in der Tat zu der professionellsten antigenpräsentierenden Zelle, die der Körper kennt (Banchereau und Steinman 1998, Bell et al. 1999) |
Ein Verweis auf die Quelle fehlt. |
|
| [7.] Dml/Fragment 031 01 - Diskussion Zuletzt bearbeitet: 2018-03-11 19:31:25 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 31, Zeilen: 3-33 |
Quelle: Regn 2002 Seite(n): 61, Zeilen: 1 ff. |
|---|---|
| EXTRAKTION VON MONONUKLEÄREN ZELLEN DES PERIPHEREN BLUTES (PBL)
Das für diese Arbeit verwendete Blut wurde entweder als Vollblut (50-100 ml), aus der Armvene von gesunden freiwilligen Spendern entnommen oder stammte von leukozytenhaltigen Präparaten („buffy coats“) des Bayerischen Roten Kreuzes. Das Blut wurde mit sterilen Einmalspritzen entnommen, denen 0.1 ml (500 IU) Heparin Novo pro 10 ml Blut beigesetzt war, um die Gerinnung des Blutes zu verhindern. Das so behandelte Blut wurde unmittelbar für die entsprechenden Experimente weiterverarbeitet. Die serologische Überprüfung aller Blutspendeproben zeigten Immunglobuline sowohl gegen AdV als auch gegen EBV. Dies wurde vom Instiut [sic] für medizinische Mikrobiologie des Max von Pettenkofer Institutes, München bestätigt. Alle folgenden im Zusammenhang mit Zellseparation und Zellkultivierung beschriebenen Vorgänge wurden unter strikt sterilen Bedingungen vorgenommen. Mononukleäre Zellen des peripheren Blutes wurden mittels Ficoll- Hypaque (spezifische Dichte = 1,078) Dichtegradientenzentrifugation isoliert. Dabei wurde das Blut zunächst im Verhältnis 1:1 mit Dulbeccos-PBS verdünnt. In 50 ml Zentrifugenröhrchen wurden 20 ml Ficollösung vorgelegt, auf die das verdünnte Blut vorsichtig mit einer 10ml Pipette überschichtet wurde. Nach 20minütiger Zentrifugation bei 800 g hat sich die mononukleäre Fraktion der Lymphozyten und Monozyten aufgrund ihrer geringeren Dichte deutlich von den Erythrozyten, den Granulozyten abgegrenzt. Während letztere durch das Ficoll- Hypaque hindurch zentrifugiert wurden, bildeten die mononukleären Zellen eine milchige Scheibe, die leicht aus dem Ficoll mit Hilfe einer sterilen Pasteurpipette aufgenommen werden konnte. Diese Zellen wurden dann in 50 ml Zentrifugenröhrchen überführt und im Verhältnis 1:1 mit Dulbeccos-PBS gewaschen und 10 Minuten bei 400 g zentrifugiert. Der Überstand abgesaugt und das Pellet nach leichtem Aufschütteln in 50ml Dulbeccos-PBS aufgenommen und 10 Minuten bei 1800 rpm zentrifugiert. Der Überstand wieder abgesaugt, das Pellet kurz aufgeschüttelt und in 10 - 20 ml RPMI-1640 Medium aufgenommen und sorgfältig durchmischt. Die Zellen wurden anschließend mit einem Hämatozytometer gezählt. |
Extraktion von mononukleären Zellen des peripheren Blutes (PBL)
Das für diese Arbeit verwendete Blut wurde entweder als Vollblut (50-100 ml), aus der Armvene von gesunden freiwilligen Spendern entnommen oder stammte von käuflich erworbenen leukozytenhaltigen Präparaten („buffy coats“) des Bayerischen Roten Kreuzes. Das Blut wurde mit sterilen Einmalspritzen entnommen, denen 0.1 ml (500 IU) heparin Novo pro 10 ml Blut beigesetzt war, um ein Gerinnen des Blutes zu verhindern. Das so behandelte Blut wurde unmittelbar für die entsprechenden Experimente weiterverarbeitet. Die serologische Überprüfung aller Blutspendeproben zeigten Immunglobuline sowohl gegen AdV als auch gegen EBV. Dies wurde vom Instiut [sic] für medizinische Mirobiologie [sic] des Max von Pettenkofer Institutes, München bestätigt. Alle folgenden im Zusammenhang mit Zellseparation und Zellkultivierung beschriebenen Vorgänge wurden unter strikt sterilen Bedingungen vorgenommen. Mononukleäre Zellen des peripheren Blutes wurden mittels Ficoll-Hypaque (spezifische Dichte = 1,078) Dichtegradientenzentrifugation isoliert. Dabei wurde das Blut zunächst im Verhältnis 1:1 mit Dulbeccos-PBS verdünnt. In 50 ml Zentrifugenröhrchen wurden 20 ml Ficollösung vorgelegt, auf die das verdünnte Blut vorsichtig mit einer 10ml Pipette überschichtet wurde. Nach 20minütigem Zentrifugieren bei 800 g hat sich die mononukleäre Fraktion der Lymphozyten und Monozyten aufgrund ihrer geringeren Dichte deutlich von den Erythrozyten, den Granulozyten abgegrenzt. Während letztere durch das Ficoll-Hypaque hindurch zentrifugiert wurden, bilden die Mononukleären Zellen einen deutlichen Ring, der leicht aus dem Ficoll mit Hilfe einer sterilen Pasteurpipette aufgenommen werden konnte. Diese Zellen wurden dann in 50 ml Zentrifugenröhrchen überführt und im Verhältnis 1:1 mit Dulbeccos-PBS gewaschen und 10 Minuten bei 400 g zentrifugiert. Danach wurde der Überstand abgesaugt und das Pellet nach leichtem Aufschütteln in 50ml Dulbeccos-PBS aufgenommen und ein weiteres Mal 10 Minuten bei 1800 rpm zentrifugiert. Danach wurde der Überstand wieder abgesaugt, das Pellet kurz aufgeschüttelt und in 10 - 20 ml RPMI-1640 Medium aufgenommen und durch mehrmaliges auf und ab pipettieren sorgfältig durchmischt. Die Zellen wurden anschließend mit einem Hämatozytometer gezählt. |
Ein Verweis auf die Quelle fehlt. |
|
| [8.] Dml/Fragment 032 01 - Diskussion Zuletzt bearbeitet: 2018-03-11 19:38:36 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 32, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 62, 63, Zeilen: 62: 1 ff.; 63: 1 ff. |
|---|---|
| ZÄHLEN VON ZELLEN
Die Anzahl lebender Zellen wurde durch Anfärben mit Trypanblau bestimmt. Lebende Zellen nehmen dabei den blauen Farbstoff nicht auf, während sich tote Zellen dunkelblau anfärben. Dazu wurden 10 μl der Zellsuspension 1:10 mit Trypanblau verdünnt. Das Gemisch wurde dann gut gemischt und vorsichtig 10μl der Suspension in die Zählkammer eines Neubauer-Hämatozytometer gefüllt. Mit Hilfe eines inversen Mikroskops wurde die Anzahl der lebenden, ungefärbten Zellen in den vier Zählquadraten, die in 16 Einzelquadraten unterteilt sind, gezählt. Die Zellkonzentration pro ml und die Gesamtzellzahl pro Probe wurde wie folgt ermittelt:
KRYOKONSERVIERUNG VON ZELLEN Die Zellen wurden in einem totalen Volumen von 1-2x107/ml in 1ml Einfriermedium pro Kryoröhrchen eingefroren. Das Einfriermedium (90 % FCS und 10 % DMSO) und die Kryogefäße wurden vor dem Einfrieren auf Eis gekühlt. Die Zellen wurden in 50- oder 15 ml Zentrifugenröhrchen mittels Zentrifugieren pellettiert, der Überstand abgesaugt, das Pellet aufgeschüttelt und auf Eis gekühlt. Anschließend wurde das kalte Einfriermedium vorsichtig zu den Zellen gegeben, wobei durch leichtes Schütteln des Röhrchens die Zellen möglichst gleichmäßig mit dem Einfriermedium durchmischt wurden. Aus dieser Zellsuspension, wurden jeweils 1ml in die zuvor ebenfalls gekühlten und beschrifteten Kryoröhrchen gefüllt. Diese wurden dann sofort in eine Spezial- Einfrierbox überführt, bei –80 °C für 24 Stunden gelagert und anschließend zur Langzeitkryokonservierung in Behälter mit flüssigen Stickstoff transferiert. AUFTAUEN VON ZELLEN Zum Auftauen von Zellen wurden 6 ml 37 °C warmes RPMI-1640 + 20 % FCS als Auftaumedium verwendet und in 15 ml-Zentrifugenröhrchen vorgelegt. |
Zählen von Zellen
Die Anzahl lebender Zellen wurde durch Anfärben mit Trypanblau bestimmt. Lebende Zellen nehmen dabei den blauen Farbstoff nicht auf, während sich tote Zellen dunkelblau anfärben. Dazu wurden 10 μl der Zellsuspension 1:10 mit Trypanblau verdünnt. Das Gemisch wurde dann mehrmals durch auf und ab pipettieren gut gemischt und dann vorsichtig 10μl der Suspension in die Zählkammer eines Neubauer-Hämatozytometer gefüllt. Mit Hilfe eines inversen Mikroskops wurde die Anzahl der lebenden, ungefärbten Zellen in den vier Zählquadraten, die in 16 Einzelquadraten unterteilt sind, gezählt. Die Zellkonzentration pro ml und die Gesamtzellzahl pro Probe wurde wie folgt ermittelt:
Kryokonservierung von Zellen Die Zellen wurden in einem totalen Volumen von 1-2 x107/ml in 1ml Einfriermedium pro Kryoröhrchen eingefroren. Das Einfriermedium (90 % FCS und 10 % DMSO) und die Kryogefäße wurden vor dem Einfrieren auf schmelzendem Eis gekühlt. Die Zellen wurden in 50- oder 15 ml Zentrifugenröhrchen mittels Zentrifugieren pellettiert, der Überstand abgesaugt, das Pellet aufgeschüttelt und ebenfalls auf schmelzendem Eis gekühlt. Anschließend wurde das kalte Einfriermedium vorsichtig zu den Zelllen [sic] gegeben, wobei durch leichtes Schütteln des Röhrchens die Zellen möglichst gleichmäßig mit dem Einfriermedium durchmischt wurden. Aus dieser Zellsuspension, die auch weiterhin auf Eis gekühlt blieb, wurden jeweils 1ml in die zuvor ebenfalls gekühlten und beschrifteten Kryoröhrchen gefüllt. Diese wurden dann sofort in eine Spezial-Einfrierbox überführt, bei –80 °C für 24 Stunden gelagert und anschließend zur Langzeitkryokonservierung in Behälter mit flüssigen Stickstoff transferiert. [Seite 63] Auftauen von Zellen Zum Auftauen von Zellen wurden 6 ml 37 °C warmes RPMI-1640 + 20 % FCS als Auftaumedium verwendet und in 15 ml-Zentrifugenröhrchen vorgelegt. |

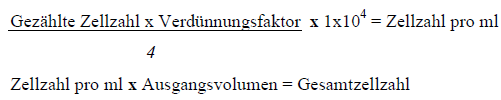
Ein Verweis auf die Quelle fehlt. |
|
| [9.] Dml/Fragment 033 01 - Diskussion Zuletzt bearbeitet: 2018-03-12 17:45:27 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 33, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 63, Zeilen: 4 ff. |
|---|---|
| Die Kryogefäße wurden direkt aus dem Behälter mit flüssigem Stickstoff entnommen und sofort für kurze Zeit in einem Wasserbad von 37 °C aufgetaut. Es wurde darauf geachtet, daß nicht der gesamte Inhalt des Gefäßes auftaut, sondern noch ein kleiner gefrorener Anteil im Innern des Gefäßes verblieb.
Ein Milliliter des Auftaumediums wurde vorsichtig mit einer Pasteurpipette in das Kryogefäß pipettiert und dabei auch das letzte Klümpchen geschmolzen. Die Zellsuspension wurde dann, mit Hilfe der Pasteurpipette tropfenweise in das 15 ml-Zentrifugenröhrchen überführt, das kontinuierlich leicht geschüttelt wurde. Die Zellen wurden dann zentrifugiert und der Überstand vorsichtig abgesaugt und in einem geeigneten Volumen von komplettem Medium aufgenommen und für weitere Experimente verwendet. KULTUR VON ADHÄRENTEN ZELLLINIEN (GHD, HUMANE FIBROBLASTEN) Die murine Fibroblasten Zelllinie L-CD40L wurde nach bereits beschriebener Methodik aufgetaut und in 6ml kompletten RPMI-1640 Medium aufgenommen und danach in 25 cm2 Zellkulturflaschen ausgelegt. Nach 24 h hatten sich ca. 60 % aller Zellen auf dem Flaschenboden angeheftet. Nach weiteren 18- 24 h war der gesamte Flaschenboden mit einem konfluenten Zellrasen bedeckt, man nennt dies Konfluenz. Das Passagieren dieser Zellen wurde wie folgt durchgeführt. Zunächst wurde das Medium abgesaugt und 5 ml PBS in die Kulturflasche gegeben. Unter kreisenden Bewegungen wurde das PBS auf den Zellen verteilt um einen Wascheffekt zu erzielen. Danach wurde das PBS abgesaugt und ca. 500 μl Versene zugegeben, ein Trypsinderivat, das die Zell-Zellkontakte lösen soll. Das Versene wurde durch Klopfen und Kreisen der Kulturflaschen gut verteilt und anschließend für 5 Min bei 37 °C inkubiert. Danach wurden die Ablösung der Zellen im Mikroskop kontrolliert. Zum vollständigen Ablösen der Zellen mußten diese vom Flaschenboden abgeklopft werden und in ca. 5- 10 ml CM aufgenommen werden. Nach einem Waschschritt um das Versene gründlich zu entfernen, wurde diese Zellsuspension dann in 75 cm2 Zellkulturflaschen transferiert und mit komplettem RPMI-1640 Medium auf ein Gesamtvolumen von 15 ml aufgefüllt. |
Die Kryogefäße wurden direkt aus dem Behälter mit flüssigem Stickstoff geholt und sofort für kurze Zeit in einem Wasserbad von 37 °C zum Auftauen gebracht. Dabei ist zu beachten, daß nicht der gesamte Inhalt des Gefäßes auftaut, sondern sich noch ein kleines gefrorenes Klümpchen im Innern des Gefäßes befindet.
Ein ml des Auftaumediums wurde nun vorsichtig mit einer Pasteurpipette in das Kryogefäß pipettiert und dabei auch das letzte Klümpchen zum Auftauen gebracht. Die Zellsuspension wurde dann, mit Hilfe der Pasteurpipette tropfenweise in das 15 ml-Zentrifugenröhrchen überführt, das währenddessen kontinuierlich leicht geschüttelt wurde. Die Zellen wurden dann zentrifugiert und der Überstand vorsichtig abgesaugt und dann in einem geeigneten Volumen von komplettem Medium aufgenommen und für weitere Experimente verwendet. Kultur von adhärenten Zellinien (L-Zellen-CD40L, Hela –Zellen, humane Fibroblasten) Die murine Fibroblasten Zellinie L-CD40L wurde nach bereits beschriebener Methodik aufgetaut und in 6ml kompletten RPMI-1640 Medium aufgenommen und danach in 25 cm2 Zellkulturflaschen ausgelegt. Nach 24 h haben sich ca. 60 % aller Zellen auf dem Flaschenboden angeheftet. Nach weiteren 18- 24 h war der gesamte Flaschenboden mit einem zusammenhängenden Zellrasen bedeckt, man nennt dies Konfluenz. Das Passagieren dieser Zellen wurde folgendermaßen durchgeführt. Zunächst wurde das Medium abgesaugt und 5 ml PBS in die Kulturflasche gegeben. Unter kreisenden Bewegungen wurde das PBS auf den Zellen verteilt um einen Wascheffekt zu erzielen. Danach wurde das PBS abgesaugt und ca. 500 μl Versene zugegeben, ein Trypsinderivat, das die Zell-Zellkontakte lösen soll. Das Versene wurde durch Klopfen und Kreisen der Kulturflaschen gut verteilt und anschließend für 5 Min bei 37 °C inkubiert. Danach wurden sich ablösende Zellen im Mikroskop kontrolliert. Zum vollständigen Ablösen der Zellen mußten diese vom Flaschenboden abgeklopft werden und in ca. 5- 10 ml CM aufgenommen werden. Diese Zellsuspension wurde dann in 75 cm2 Zellkulturflaschen transferriert [sic] und mit komplettem RPMI-1640 Medium auf ein Gesamtvolumen von 15 ml aufgefüllt. |
Ein Verweis auf die Quelle fehlt. |
|
| [10.] Dml/Fragment 034 01 - Diskussion Zuletzt bearbeitet: 2018-03-16 19:03:54 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 34, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 64, 66, 67, Zeilen: 64: 1 ff.; 66: letzter Abschnitt; 67: 1 ff. |
|---|---|
| KULTUR VON DENDRITISCHEN ZELLEN (DCS)
PBL wurden in serumfreien RPMI-1640 in einer Konzentration von 2,5x106 pro ml in 6-Well-Platten ausgesät, mit einer Gesamtzellzahl von 5x106 Zellen pro Well. Die Platten wurden dann für zwei Stunden bei 37 °C und 5 % CO2 inkubiert. Nach Absetzen der adhärenten Zellfraktion wurde der Überstand vorsichtig mit einer sterilen Pasteurpipette von der adhärenten Zellschicht heruntergewaschen, so dass das gesamte Kulturmedium mit den nicht-adhärenten Zellen entfernt wurde, diese wurden für den späteren Gebrauch aufgehoben. Die adhärenten Zellen wurden weiter kultiviert unter Zugabe von 2 ml serumfreien Medium (X-VIVO-15), supplementiert mit 2 mM L-Glutamin und Penicillin-Streptomycin (100 U/ml und 100 μg/ml), sowie 800 IU/ml rekombinantem humanes GM-CSF und 500 IU/ml rekombinantem human IL-4. Nach sieben Tagen in Kultur wurde frisches rhGM-CSF (800 IU/ml) dem Kulturmedium zugegeben und nach 10-12 Tagen Zellkultur [sic] die unreifen DCs geerntet und für weiterführende Experimente verwendet. PHÄNOTYPISCHE ANALYSE VON DCS Zur Charakterisierung von DCs mittels direkter Immunfluoreszenz wurden die folgenden monoklonalen Antikörper verwendet, die entweder mit Fluoresceinisothiocyanat (FITC) oder Phycoerythrin (PE) direktmarkiert waren: CD-1a (FITC,-oder PE-konjugiert), CD-11c-PE, CD-11a –FITC, CD-123-PE, CD-25-FITC, CD-40 (FITC,-oder PE-konjugiert), CD-80, CD-86, (FITC,-oder PE-konjugiert), CD-51-PE, HLA-ABC und HLA-DR (FITC,-oder PE-onjugiert), CD-19-FITC, CD-83-PE und CD-14-PE BESTIMMUNG DER ENDOZYTOTISCHEN KAPAZITÄT VON DCS Die Mannoserezeptor vermittelte Endozytose stellt experimentell ein Maß dar, für die Fähigkeit DCs Moleküle aufzunehmen, zu prozessieren und zu präsentieren. Aufgrund multipler Kohlenhydratbindungsdomänen des Mannoserezeptors, sind DCs befähigt große Mengen an Glykoproteinen zu binden und zu internalisieren. Im Gegensatz zu Fc-Rezeptoren, die zusammen mit ihren Liganden degradiert werden, entläßt der Mannoserezeptor seine Liganden bei einem endosomalen pH in endosomale Vesikel und wird dabei selbst wieder verwendet. |
Kultur von Dendritischen Zellen (DCs)
PBL wurden in serumfreien RPMI-1640 in einer Konzentration von 2,5x106 pro ml in 6-Well-Platten ausgesät, mit einer Gesamtzellzahl von 5x106 Zellen pro Plattenvertiefung. Die Platten wurden dann für zwei Stunden bei 37 °C und 5 % CO2 inkubiert. In der Zwischenzeit hat sich eine adhärente Zellfraktion, von einer nicht-adhärenten Zellfraktion abgesetzt, die vorsichtig mit einer sterilen Pasteurpipette von der adhärenten Zellschicht heruntergewaschen und für späteren Gebrauch aufgehoben wurde. Dabei mußte beachtet werden, daß das gesamte Kulturmedium mit den nicht-adhärenten Zellen entfernt wurde. Die adhärenten Zellen wurden weiter kultiviert unter Zugabe von 2 ml serumfreien Medium (X-VIVO-15), supplementiert mit 2 mM L-Glutamin und Penicillin-Streptomycin (100 U/ml und 100 μg/ml), sowie 800 IU/ml rekombinantem humanes GM-CSF und 500 IU/ml rekombinantem humanes IL-4. Nach sieben Tagen in Kultur wurde frisches rhGM-CSF (800 IU/ml) dem Kulturmedium zugegeben und nach 10-12 Tagen Zellkultur konnten die unreifen DCs geerntet und für weiterführende Experimente verwendet werden. [...] Phänotypische Analyse von DCs Zur Charakterisierung von DCs mittels direkter Immunfluoreszenz wurden die folgenden monoklonalen Antikörper verwendet, die entweder mit Fluorescein-isothiocyanate (FITC) oder Phycoerythrin (PE) direktmarkiert waren: CD1a (FITC,-oder PE-konjugiert), CD11c-PE, CD11a –FITC, CD123-PE, CD25-FITC, CD40 (FITC,-oder PE-konjugiert), CD80, CD86, (FITC,-oder PE-konjugiert), CD51-PE, HLA-ABC und HLA-DR (FITC,-oder PE-konjugiert), CD19-FITC, CD83-PE und CD14-PE [Seite 66] Bestimmung der endozytotischen Kapazität von DCs mittels FITC-Dextran Die Mannoserezeptor vermittelte Endozytose stellt experimentell ein Maß für die Funktionalität von DCs dar. Aufgrund multipler Kohlenhydratbindungsdomänen des Mannoserezeptors, sind DCs befähigt große Mengen an Glykoproteinen zu binden und zu internalisieren. Im Gegensatz zu Fc- [Seite 67] Rezeptoren, die zusammen mit ihren Liganden degradiert werden, entläßt der Mannoserezeptor seine Liganden bei einem endosomalen pH in endosomale Vesikel und wird dabei selbst wiederverwendet. |
Ein Verweis auf die Quelle fehlt. |
|
| [11.] Dml/Fragment 035 01 - Diskussion Zuletzt bearbeitet: 2018-03-11 19:04:46 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 35, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 66, 67, Zeilen: 66: 1 ff.; 67: 3 ff. |
|---|---|
| [Daher kann eine] große Menge von verschiedenen Liganden gebunden und aufgenommen werden, bei einer relativ niedrigen Menge an Rezeptoren.
FITC-konjugiertes Dextran (MW:70000) wurde mit serumfreien Medium zu einer Endkonzentration von 1 mg/ml verdünnt. DCs wurden in einer Konzentration von 1x105 pro Test in 100 μl X-VIVO 15 eingestellt und mit 100 μl der FITC-Dextranlösung versetzt. Das Gesamtvolumen betrug dabei 200 μl und wurde bei 37 °C inkubiert. Es wurden doppelte Testansätze vorbereitet und bei einer Kinetik von 30-180 Min. wurden die Zellen aus dem Inkubator entnommen und 2 x mit kaltem PBS gewaschen und in 500 μl FACS-Puffer aufgenommen und die Menge an aufgenommene FITC-Dextran im FACS Gerät gemessen. Unbehandelte DCs dienten dabei als Negativkontrolle. DURCHFLUßZYTOMETRISCHE ANALYSEN Der Fluoreszenzaktivierte Zellsorter (fluorescence-activated cell sorter = FACS ) kann nicht nur Zellen nach Größe und Granularität einordnen, sondern auch zählen und gemäß ihrer Oberflächenmarkierung auftrennen und quantifizieren. Monoklonale Antikörper gegen verschiedene Oberflächenproteine werden mit Fluoreszenzfarbstoffen gekoppelt, um bestimmte Zellen in einer gemischten Population detektieren zu können. Das Gemisch der markierten Zellen wird angesaugt und anschließend durch eine Kapillare gedrückt. Dadurch entsteht ein feiner Flüssigkeitsstrahl mit vereinzelten Zellen, die sich in bestimmten Abständen befinden. Dieser Flüssigkeitsstrahl passiert einen Laserstrahl, dabei kommt es an den Zellen zu einer Lichtstreuung und die Farbstoffmoleküle, die über die monoklonalen Antikörper an die Zelle gebunden sind werden zur Fluoreszenz angeregt. Empfindliche Photodetektoren messen sowohl das gestreute als auch das emittierte Licht, wobei ersteres Informationen über Größe und Granularität der Zelle liefert, die Fluoreszenz ermöglicht dagegen Aussagen über die Bindung der monoklonalen Antikörper und damit über die Expression der Oberflächenproteine auf jeder untersuchten Zelle. |
[Seite 67]
Daher können eine große Menge von verschiedenen Liganden gebunden und aufgenommen werden, bei einer relativ niedrigen Menge an Rezeptoren. FITC-konjugiertes Dextran (MW:70000) wurde mit serumfreien Medium zu einer Endkonzentration von 1 mg/ml verdünnt. DCs wurden in einer Konzentration von 1x105 pro Test in 100 μl X-VIVO 15 eingestellt und mit 100 μl der FITC-Dextranlösung versetzt. Das Gesamtvolumen betrug dabei 200 μl und wurde bei 37 °C inkubiert. Es wurden doppelte Testansätze vorbereitet und bei einer Kinetik von von 30-180 Min. wurden die Zellen aus dem Inkubator entnommen und 2 x mit kaltem PBS gewaschen und in 500 μl FACS-Buffer aufgenommen und die Menge an aufgenommene FITC-Dextran im FACS Gerät gemessen. Unbehandelte DCs dienten dabei als Negativkontrolle. [Seite 66] Durchflußzytometrische-Analyse Der Fluoreszenzaktivierte Zellsorter (fluorescence-activated cell sorter = FACS ) ist ein Gerät, daß [sic] nicht nur Zellen nach Größe und Granularität einordnen kann, sondern auch zählen und gemäß ihrer Oberflächenmarkierung auftrennen und quantifizieren kann. Monoklonale Antikörper gegen verschiedene Oberflächenproteine werden mit Fluoreszenzfarbstoffen gekoppelt, um bestimmte Zellen in einer gemischten Population detektieren zu können. Das Gemisch der markierten Zellen wird angesaugt und anschließend durch eine Kapillare gedrückt. Dadurch entsteht ein feiner Flüssigkeitsstrahl mit vereinzelten Zellen, die sich in bestimmten Abständen befinden. Dieser Flüssigkeitsstrahl passiert einen Laserstrahl, dabei kommt es an den Zellen zu einer Lichtstreuung und die Farbstoffmoleküle, die über die monoklonalen Antikörper an die Zelle gebunden sind werden zur Fluoreszenz angeregt. Empfindliche Photodetektoren messen sowohl das gestreute als auch das emittierte Licht, wobei ersteres Informationen über Größe und Granularität der Zelle liefert, die Fluoreszenz ermöglicht dagegen Aussagen über die Bindung der monoklonalen Antikörper und damit über die Expression der Oberflächenproteine auf jeder untersuchten Zelle. |
Ein Verweis auf die Quelle fehlt. |
|
| [12.] Dml/Fragment 036 01 - Diskussion Zuletzt bearbeitet: 2018-03-14 17:05:00 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 36, Zeilen: 1-24 |
Quelle: Regn 2002 Seite(n): 65, 75, Zeilen: 65: 1 ff.; 75: 11 ff. |
|---|---|
| FÄRBUNG MIT FLOURESZENZMARKIERTEN [sic] MONOKLONALEN ANTIKÖRPERN
Das Anfärben der DCs mittels direkter 2-Farben-Immunfluoreszenzmarkierung wurde wie folgt durchgeführt: Ernten der Zellen durch sorgfältiges Abpipettieren der Zellen vom Plattenboden unter Zuhilfenahme einer Plastikpastette und transferieren in ein Zentrifugenröhrchen. Die Zellen wurden 2 mal mit FACS-Puffer (PBS + 2 % FCS) gewaschen. Aufnehmen des Pellets in einer geeigneten Menge von FACS Puffer und verteilen von jeweils 100 μl der Zellsuspension in Durchflusszytometrie- Teströhrchen (FACS-tubes) pro Färbung. Zugabe von jeweils 2-3 μl des Fluoresceinisothiocyanat (FITC) und des Phycoerythrin (PE)-konjugierten monoklonalen Antikörpers (mAb). Inkubieren der Teströhrchen im Dunkeln für 15 Minuten bei Raumtemperatur. Zugabe von 800 μl FACS-Puffer und zentrifugieren für 5 Min. bei 400 g. Verwerfen des Überstandes und erneute Zugabe von 800 μl FACS-Puffer. Zentrifugieren für 5 Min. bei 400 g Aufnahme des Pellets in 500 μl FACS-Puffer und Analyse der Oberflächenmarker in der Durchflußzytometrie IMMUNPHÄNOTYPISIERUNG DER PROLIFERIERENDEN AUTOLOGEN UND ALLOGENEN T-ZELLEN Zur Bestimmung der Oberflächenexpression spezifischer T-Zellmarker wurden die T-Zellen mit verschiedenen Kombinationen von FITC-oder PE-konjugierten monoklonalen Antikörpern inkubiert und anschließend in der Durchflusszytometrie ausgewertet. Eingesetzt wurden dazu folgende Antikörper: CD-3, CD-8, CD-4, CD-56. |
Färbung mit fluoreszenzmarkierten monoklonalen Antikörpern
Das Anfärben der DCs mittels direkter 2-Farben-Immunfluoreszenzmarkierung wurde wie folgt durchgeführt:
[Seite 75] Immunphänotypisierung der autologen und allogenen T-Zellen Zur Bestimmung der Oberflächenexpression spezifischer T-Zellmarker wurden die T-Zellen mit verschiedenen Kombinationen von FITC-oder PE-konjugierten monoklonalen Antikörpern inkubiert und anschließend in der FACS-Analyse ausgewertet. Eingesetzt wurden dazu folgende Antikörper: CD3, CD8, CD4, CD56. |
Ein Verweis auf die Quelle fehlt. |
|
| [13.] Dml/Fragment 038 20 - Diskussion Zuletzt bearbeitet: 2018-03-12 20:37:57 WiseWoman | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 38, Zeilen: 20-30 |
Quelle: Regn 2002 Seite(n): 69, 70, Zeilen: 69: letzter Abschnitt; 70: 1 ff. |
|---|---|
| TRANSFEKTION VON DCS MIT DOTAP
Die gewonnen [sic] Poly-RNA wurde in der benötigten Menge an infektiösen Partikel mit 50 μl OptiMEM in einem 1,5ml Eppendorf -Reaktionsgefäß verdünnt. In einem zweiten 1,5ml Eppendorf-Reaktionsgefäß wurden 1.25 μg des kationischen Lipids DOTAP in 50 μl OptiMem gelöst. Anschließend wurden das Lipidgemisch vorsichtig mit einer Eppendorfpipette zu dem RNA Gemisch [sic] gegeben wurde, um Scherkräfte zu vermeiden. Durch leichtes Klopfen gegen die Wand des Reaktionsgefäßes wurde eine Vermischung beider Lösungen erzielt. Anschließend wurde das Gemisch für 15 Minuten bei Raumtemperatur inkubiert. In der Zwischenzeit wurden die DCs geerntet, gewaschen und mit einer Konzentration von 1x106 Zellen in 100 μl OptiMEM [eingestellt.] |
Adenovirale Transfektion von DCs mit Lipofectamin
Das Virus wurde in der benötigten Menge an infektiösen Partikel mit 50 μl OptiMEM in einem 1,5ml Eppendorf -Reaktionsgefäß verdünnt. In einem zweiten 1,5ml Eppendorf-Reaktionsgefäß wurden 1.25 μg des kationischen Lipids Lipofectamin in 50 μl OptiMem gelöst. [Seite 70] Anschließend wurden beide Lösungen miteinander vereinigt indem das Lipidgemisch vorsichtig mit einer Eppendorfpipette zu dem Virusgemisch gegeben wurde, um Scherkräfte zu vermeiden. Durch leichtes Klopfen gegen die Wand des Reaktionsgefäßes wurde eine Vermischung beider Lösungen erzielt. Anschließend wurde das Gemisch für 15 Minuten bei Raumtemperatur inkubiert. In der Zwischenzeit wurden die DCs geerntet, gewaschen und mit einer Konzentration von 1x106 Zellen in 100 μl OptiMEM eingestellt. |
Ein Verweis auf die Quelle fehlt. |
|
| [14.] Dml/Fragment 039 01 - Diskussion Zuletzt bearbeitet: 2018-03-12 17:17:03 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 39, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 68, 70, 73, Zeilen: 68: 1 ff.; 70: 6 ff.; 73: 8 ff. |
|---|---|
| Nach Ende der 15 minütigen Inkubationszeit wurden die Zellen mit dem RNA-Lipidgemisch vereinigt und für 60 Minuten bei 37 °C inkubiert. Nach der Inkubationszeit wurden die Zellen mit PBS gewaschen und anschließend in 2ml X-VIVO 15 unter Zugabe von 400 U/ml GM-CSF und 200 U/ml TNF-α aufgenommen und in ein Well einer 24-Well- Platte transferiert.
HERSTELLUNG EBV-TRANSFORMIERTER B-ZELL- LINIEN (LCL) PBL wurden in komplettem RPMI-1640 zu einer Konzentration von 5x106 Zellen pro Well in 24-Well Zellkulturplatten angesetzt. Die transformierenden Eigenschaften des EBV-haltigen B-95-8 Überstandes wurden durch limitierende Verdünnungsreihen zwischen 100 und 1000 μl an seronegativen und seropositiven Spendern getestet und eine Menge von 300- 500 μl des Überstandes wurde als ideal für eine ausreichende Transfektion erachtet. Die Zellen wurde mit dem Überstand für eine Stunde bei 37° C inkubiert. Anschließend wurde 1 μg/ml Cyclosporin A hinzugegeben, dies führt zu dem sogenannten „T-Cell-knockout“ und verhindert eventuelle Abwehrreaktionen. Nach 7 Tagen in Kultur wurden die Zellen mit frischem Medium versorgt und nach ca. vier Wochen waren die Zell-Linien etabliert und konnten in 25 cm2 Zellkulturflaschen expandiert werden. Nach Expansion in 75 cm2 Zellkulturflaschen wurden in regelmäßigen Abständen Aliquots der Zellen zur Langzeitlagerung in flüssigen Stickstoff kryokonserviert. ASSAYS GEMISCHTE LYMPHOZYTEN REAKTION (MLR = MIXED LYMPHOZYTE [sic] REACTION) Die gemischte Lymphozytenreaktion (MLR= „mixed lymphocyte reaction“) ist ein Experiment, das ursprünglich als Modell für die Transplantatabstoßung entwickelt wurde. Bestrahlte Lymphozyten eines potentiellen Spenders (=Stimulatorzellen) wurde mit unbestrahlten Lymphozyten eines Empfängers (= Responder-T-Zellen) zusammengemischt. Sind Spender und Empfänger in ihren HLA-Antigenen unterschiedlich, dann werden die T- Zellen in der Lymphozytenkultur des Empfängers zur Proliferation angeregt, weil sie gegen diese HLA-Antigene reagieren und entwickeln sich durch die resultierende Zytokinproduktion zu Killerzellen. Die MLR kann daher als Maß für die immunstimulatorische Kapazität [von APCs, wie DCs oder LCLs eingesetzt werden.] |
[Seite 70]
Nach Ende der 15 minütigen Inkubationszeit wurden die Zellen mit dem Virus-Lipidgemisch vereinigt und für 60 Minuten bei 37 °C inkubiert. Nach der Inkubationszeit wurden die Zellen mit PBS gewaschen und anschließend in 2ml X-VIVO 15 unter Zugabe von 400 U/ml GMCSF und 200 U/ml TNF-α aufgenommen und in ein Well einer 24-Well- Platte transferiert. [Seite 68] Herstellung EBV-transformierter B-Zell- Linien (LCL) PBL wurden in komplettem RPMI-1640 zu einer Konzentration von 5x106 Zellen pro Well in 24-Well Zellkulturplatten ausgesät. Die transformierenden Eigenschaften des EBV-haltigen B-95-8 Überstandes wurden durch limitierende Verdünnungsreihen zwischen 100 und 1000 μl an seronegativen und seropositiven Spendern getestet und eine Menge von 300- 500 μl des Überstandes wurde als ideal erachtet und zu den Zellen gegeben, und wurde dann für eine Stunde bei 37° C inkubiert. Anschließend wurde 1 μg/ml Cyclosporine A hinzugegeben. Nach 7 Tagen in Kultur wurden die Zellen mit frischem Medium versorgt und nach ca. vier Wochen waren die Zell-Linien etabliert und konnten in 25 cm2 Zellkulturflaschen expandiert werden. Nach Expansion in 75 cm2 Zellkulturflaschen wurden in regelmäßigen Abständen Aliquots der Zellen zur Langzeitlagerung in flüssigen Stickstoff kryokonserviert. [Seite 73] Gemischte Lymphozyten Reaktion (MLR = mixed lymphozyte [sic] reaction) Die gemischte Lymphozytenreaktion (MLR= „mixed lymphozyte [sic] reaction“) ist ein Experiment, das ursprünglich als Modell für die Transplantatabstoßung entwickelt wurde. Bestrahlte Lymphozyten eines potentiellen Spenders (=Stimulatorzellen) wurde mit unbestrahlten Lymphozyten eines Empfängers (= Responder-T-Zellen) zusammengemischt. Sind Spender und Empfänger in ihren HLA-Antigenen unterschiedlich, dann werden die T- Zellen in der Lymphozytenkultur des Empfängers zur Proliferation angeregt, weil sie gegen diese HLA-Antigene reagieren und entwickeln sich durch die resultierende Zytokinproduktion zu Killerzellen. Die MLR kann daher als Maß für die immunstimulatorische Kapazität von APCs, wie DCs oder LCLs eingesetzt werden. |
Ein Verweis auf die Quelle fehlt. |
|
| [15.] Dml/Fragment 040 01 - Diskussion Zuletzt bearbeitet: 2018-03-16 19:15:20 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 40, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 73, 74, Zeilen: 73: 17 ff.; 74: 1 ff. |
|---|---|
| In autologen und allogenen Testsystemen sollte die Kapazität RNA-infizierter DCs untersucht werden die Proliferation von T-Zellen zu induzieren.
Die Vorbereitung ist bei allogenen und autologen MLR identisch: Aus frischem Spenderblut isolierte PBL wurden zur Aufreinigung von T-Zellen verwendet. Dazu wurde das MACS Pan T Cell Isolation Kit der Firma Miltenyi Biotec verwendet. Nach der Isolation der PBL über den Ficoll Gradienten wurden die Zellen gewaschen und gezählt. Jeweils 1x107 Zellen wurden in 80μl MACS-Puffer aufgenommen und mit 20μl des Hapten-Antibody-Cocktail (enthält eine Mixtur aus monoklonalen haptenkonjugierten Antikörpern: CD11b, CD19, CD36 und CD56) vermischt und bei 6-12°C in einem Eis-Wassergemisch für 10 Minuten inkubiert. Nach der Inkubationszeit wurden die Zellen 2 mal mit MACS-Puffer gewaschen und anschließend erneut in 80μl MACS-Puffer aufgenommen und mit 20μl der MACS Anti-Hapten Microbeads (enthält magnetische Teilchen, die an monoklonale anti-Hapten Antikörper gebunden sind) versetzt und gut durchmischt. Die Zellsuspension wurde dann für weitere 15 min bei 6-12°C in dem Eis-Wassergemisch inkubiert. Danach wurden die Zellen 1 mal gewaschen und in einem Volumen von 500μl MACS-Puffer aufgenommen. Zur T-Zell-Depletion wurden mit feiner Stahlwolle (oder Kügelchen) gefüllte LS+/VS+-Säulen an einem Magneten (MidiMACS, VarioMACS oder SuperMACS) befestigt und mit 3 ml MACS-Puffer befeuchtet. Anschließend wurde die Zellsuspension auf die Säule gegeben und die durchlaufende Fraktion in einem Röhrchen gesammelt. Alle Zellen, die nicht den T-Zellen entsprachen, blieben aufgrund der Markierung mit den Antikörper-Microbeads in der Säule hängen, während die unmarkierte negative Fraktion hochangereicherte CD3+ T-Zellen enthielt. Um eine möglichst hohe Ausbeute an separierten T-Zellen von der Säule zu gewinnen, wurde diese noch viermal mit 3 ml MACS-Puffer gewaschen. Die Eluate wurden vereinigt, zentrifugiert, noch einmal mit PBS gewaschen und schließlich in einem entsprechenden Volumen von komplettem Medium aufgenommen, gezählt und für weitere Analyse weiterverwendet. Die Reinheit der T-Zellen wurde durch Markierung mit einem anti-CD3-Antikörper in der Durchflusszytometrie evaluiert. Für den Ansatz in der MLR wurden die T-Zellen auf 1x106/ml eingestellt und pro 100μl in 96-Well-Rundbodenplatten gesät. |
In autologen und allogenen Testsystemen sollte die Kapazität AdV-infizierter DCs und LCLs untersucht werden die Proliferation von T-Zellen zu induzieren. Aus frischem Spenderblut isolierte PBL wurden zur Aufreinigung von T-Zellen verwendet. Dazu wurde das MACS Pan T Cell Isolation Kit der Firma Miltenyi Biotec verwendet. Nach der Isolation der PBL über den Ficoll Gradienten wurden die Zellen gewaschen und gezählt. Jeweils 1x107 Zellen wurden in 80μl MACS-Buffer aufgenommen und mit 20μl des Hapten-Antibody-Cocktail (enthält eine Mixtur aus monoklonalen haptenkonjugierten Antikörpern: CD11b, CD19, CD36 und CD56) vermischt und bei 6-12°C in einem Eis-Wassergemisch für 10 Minuten inkubiert. Nach der Inkubationszeit wurden die Zellen 2 mal mit MACS-Buffer gewaschen und anschließend erneut in 80μl MACS-Buffer aufgenommen und mit 20μl der MACS Anti-Hapten Microbeads (enthält magnetische Teilchen, die an monoklonale anti-Hapten Antikörper gebunden sind) versetzt und gut durchmischt. Die Zellsuspension wurde dann für weitere 15 min bei 6-12°C in dem Eis-Wassergemisch inkubiert. Danach wurden die Zellen 1 mal gewaschen und in einem Volumen von 500μl MACS-Buffer aufgenommen.
Zur T-Zell-Depletion wurden mit feiner Stahlwolle (oder Kügelchen) gefüllte LS+/VS+-Säulen an einem Magneten (MidiMACS, VarioMACS oder SuperMACS) befestigt und mit 3 [Seite 74] ml MACS-Buffer befeuchtet. Anschließend wurde die Zellsuspension auf die Säule gegeben und die durchlaufende Fraktion in einem Röhrchen gesammelt. Alle Nicht T-Zellen blieben aufgrund der Markierung mit den Antikörper-Microbeads in der Säule hängen, während die unmarkierte negative Fraktion hochangereicherte CD3+ T-Zellen enthielt. Um eine möglichst hohe Ausbeute an separierten T-Zellen von der Säule zu gewinnen wurde diese noch viermal mit 3 ml MACS-Buffer gewaschen. Die Eluate wurden vereinigt, zentrifugiert, noch einmal mit PBS gewaschen und schließlich in einem entsprechenden Volumen von komplettem Medium aufgenommen, gezählt und für weitere Analyse weiterverwendet. Die Reinheit der T-Zellen wurde durch Markierung mit einem anti-CD3-Antikörper in der FACS-Analyse evaluiert. Für den Ansatz in der MLR wurden die T-Zellen auf 1x106/ml eingestellt und pro 100μl in 96-Well-Rundbodenplatten gesät. |
Ein Verweis auf die Quelle fehlt. |
|
| [16.] Dml/Fragment 041 01 - Diskussion Zuletzt bearbeitet: 2018-03-14 17:19:11 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 41, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 74, 75, Zeilen: 74: 12 ff., 75: 1 ff. |
|---|---|
| [Die] antigenpräsentierenden Stimulatorzellen (DCs oder LCLs) wurden zunächst bestrahlt (30 gy für DCs und 45 gy für LCLs) und in verschiedenen Ratios in dreier Gruppen (Triplikate) zu den T-Zellen gegeben. Aufgrund der individuellen Unterschiede in der Reinheit der verschiedenen DC-Kulturen wurden die DCs immer sehr genau und sorgfältig auf die entsprechende Zellkonzentrationen eingestellt. Dabei wurde zunächst die Quantität der DCs im FACS evaluiert und anschließen [sic] die deutlich größeren Zellen gezählt [sic] während die kleineren Lymphozyten ignoriert wurden. Da LCLs relativ homogene Zellsuspensionskulturen darstellen, war hier das Einstellen auf die jeweilige Zellzahl einfacher. Die Endkonzentration in jedem Well betrug 200μl. Jeweils drei Wells wurden mit T-Zellen alleine als negative Kontrolle versehen. Nach 6 Tagen in Kultur wurde 1 μCi von Methyl-[3H]Thymidin der Kultur zugegeben und für weitere 18 Stunden inkubiert. Nur sich replizierende T-Zellen können das Methyl- [3H]Thymidin in ihre DNA einbauen. Nach Ende der Inkubationszeit wurden die [3H]Thymidin –markierten Zellen in den 96-Well-Rundbodenplatten mit Hilfe eines FilterMate TM Cell Harvester auf speziell entwickelte UniFilter-Platten transferiert und die Aufnahme des Thymidins wurde als emittierende Gamma-Strahlung im Liquid-Scintillations-Verfahren mit Hilfe eines Packard Topcounters quantifiziert. Als Maß für die Proliferation wurden counts per minute (CPM) angegeben, welche die Menge des eingebauten Thymidins repräsentieren. Die proliferierenden T-Zellen in der MLR wurden in der FACS –Analyse phänotypisiert, dabei wurden die oben beschriebenen Antikörper eingesetzt.
Unreife dendritische Zellen sind in der Lage exogene Antigene aufzunehmen und sie den T-Zellen zu präsentieren. Um diese Fähigkeit der DCs zu testen wurden RNA-infizierte DCs oder nicht infizierte DCs in komplettem Medium resuspendiert, das 20mg/ml rekombinantes Tetanus Toxoid (TT) Fragment C (Chiron Behring, Marburg) enthielt oder ohne Zusatz war. Nach 24 Stunden wurden die Zellen bestrahlt (30 Gy) und in Triplikaten zu den aufgereinigten T-Zellen (1x105/ Well) in verschiedenen Stimulator:T-Zell Ratios in 96-Well Rundbodenplatten gegeben. Nach 5 Tagen in Kultur wurde 1 μCi Methyl- [3H]Thymidin der Kultur zugegeben und für weitere 18 Stunden inkubiert. Danach wurde dann wie bereits beschrieben der Einbau des Thymidins in die proliferierenden Zellen quantifiziert. |
Die antigenpräsentierenden Stimulatorzellen (DCs oder LCLs) wurden zunächst bestrahlt (30 gy für DCs und 45 gy für LCLs) und in verschiedenen Ratios in dreier Gruppen (Triplicates) zu den T-Zellen gegeben. Aufgrund der individuellen Unterschiede in der Reinheit der verschiedenen DC-Kulturen wurden die DCs immer sehr genau und sorgfältig auf die entsprechende Zellkonzentrationen eingestellt. Dabei wurde zunächst die Quantität der DCs im FACS evaluiert und anschließend nur die großen runden Zellen gezählt, kleine Lymphozyten wurden dabei ignoriert. Da LCLs relativ homogene Zellsuspensionskulturen darstellen war hier das Einstellen auf die jeweilige Zellzahl einfacher. Die Endkonzentration in jedem Well betrug 200μl. Jeweils drei Wells wurden mit T-Zellen alleine als negative Kontrolle versehen. Nach 6 Tagen in Kultur wurde 1 μCi von Methyl-[3H]Thymidin der Kultur zugegeben und für weitere 18 Stunden inkubiert. Nur sich replizierende T-Zellen können das Methyl-[3H]Thymidin in ihre DNA einbauen. Nach Ende der Inkubationszeit wurden die [3H]Thymidin –markierten Zellen in den 96-Well-Rundbodenplatten mit Hilfe eines FilterMate TM Cell Harvester auf speziell entwickelte UniFilter-Platten transferiert und die Aufnahme des Thymidins wurde als emittierende β-Strahlung im Liquid-Scintillations-Verfahren mit Hilfe eines Packard Topcounters quantifiziert. Als Maß für die Proliferation wurden counts per minute (CPM) angegeben, welche die Menge des eingebauten Thymidins repräsentieren. Die proliferierenden T-Zellen in der MLR wurden in der FACS –Analyse phänotypisiert, dabei wurden monoklonale Antikörper, die gegen CD3, CD4, CD8 und CD56 gerichtet waren verwendet.
[Seite 75] Autologe MLR Unreife Dendritische Zellen sind in der Lage exogene Antigene aufzunehmen und sie den T-Zellen zu präsentieren. Um diese Fähigkeit der DCs zu testen wurden AdV-infizierte DCs oder nicht infizierte DCs in komplettem Medium resuspendiert, das 20mg/ml rekombinantes tetanus toxoid (TT) Fragment C (Chiron Behring, Marburg) enthielt oder ohne Zusatz war. Nach 24 Stunden wurden die Zellen bestrahlt (30 Gy) und in Triplikaten zu den aufgereinigten T-Zellen (1x105/ Well) in verschiedenen Stimulator:T-Zell Ratios in 96-Well Rundbodenplatten gegeben. Nach 5 Tagen in Kultur wurde 1 μCi Methyl-[3H]Thymidin der Kultur zugegeben und für weitere 18 Stunden inkubiert. Danach wurde dann wie bereits beschrieben der Einbau des Thymidins in die proliferierenden Zellen quantifiziert. |
Ein Verweis auf die Quelle fehlt. |
|
| [17.] Dml/Fragment 042 04 - Diskussion Zuletzt bearbeitet: 2018-03-16 16:38:25 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 42, Zeilen: 4-32 |
Quelle: Regn 2002 Seite(n): 77, Zeilen: 1 ff. |
|---|---|
| STANDARD CHROMFREISETZUNGS TEST (CHROMIUM-51 RELEASE ASSAYS)
Allgmeine [sic] Vorbereitungen zum Chromfreisetzungs Test: Autologe dendritische Zellen wurden aus frischem Blut oder von eingefrorenen PBL von „buffycoats“ 14 Tage vor der Durchführung des Assays wie beschrieben generiert. Dabei mußten die Menge [sic] so kalkuliert werden, daß folgende Zielzellen hergestellt werden konnten:
In der gleichen Weise wurden allogene dendritische Zellen von HLA-nicht identischen Spendern oder von „buffycoats“ mit der Annahme nicht übereinstimmender HLA-Typen generiert. Darüber hinaus dienten autologe LCLs, allogene LCLs und die NK-sensitive Zell-Linie HSB2 als weitere Zielzellen. Der Chromfreisetzungs Test dient zur Überprüfung der spezifischen Zellvermittelnden Zytotoxizität. Die CTLs wurden geerntet und in einer Konzentration von 2x106 Zellen /ml in komplettem RPMI aufgenommen. 96 Well Spitzbodenplatten wurden von Reihe B bis D mit jeweils 100μl komplettem RPMI pro Well gefüllt. In die oberste Reihe A, wurden 200 μl der CTL-Suspension gefüllt. Mit Hilfe einer 12-Kanal Multipette wurden dann jeweils 100 μl, angefangen bei Reihe A in die jeweils tiefer liegende Reihe pipettiert. Bei jedem Transferschritt wurden dann Zellsuspension und Medium sorgfältig durch zahlreiches Auf- und Abpipettieren durchmischt um eine gleichmäßige Verteilung zu erreichen. Bei Reihe D schließlich wurden 100 μl verworfen, da hier die Verdünnungskaskade beendet wurde. Die Reihen E und F waren für die HLA-Inhibitionsexperimente vorgesehen. In Reihe G wurden 100μl Medium und später die Chrom markierten Zielzellen für die Ermittlung der spontanen Chromfreisetzung gegeben und in Reihe H wurde schließlich das Detergenz Triton-X zugegeben, um die maximalen Freisetzung des gespeicherten Chroms zu bestimmen. |
Vorbereitung der Zielzellen für den Zytotoxizitäts-Assay
Autologe Dendritische Zellen wurden aus frischem Blut oder von eingefrorenen PBL von „buffycoats“ 14 Tage vor der Durchführung des Assays wie beschrieben generiert. Dabei mußten die Mengen so kalkuliert werden, daß folgende Zielzellen hergestellt werden konnten:
Standard Chromfreisetzungs Test („chromium-51 release assays“) oder Test zur Überprüfung der spezifischen Zell-vermittelnden Zytotoxizität Die ZTL wurden geerntet und in einer Konzentration von 2x106 Zellen /ml in komplettem RPMI aufgenommen. 96 Well Spitzbodenplatten wurden von Reihe B bis D mit jeweils 100μl komplettem RPMI pro Well gefüllt. In die oberste Reihe A, wurden 200 μl der ZTL-Suspension gefüllt. Mit Hilfe einer 12-Kanal Multipette wurden dann jeweils 100 μl, angefangen bei Reihe A in die jeweils tiefer liegende Reihe pipettiert. Bei jedem Transferschritt wurden dann Zellsuspension und Medium sorgfältig durch zahlreiches Auf- und Abpipettieren durchmischt um eine gleichmäßige Verteilung zu erreichen. Bei Reihe D schließlich wurden 100 μl verworfen, da hier die Verdünnungskaskade beendet wurde. Die Reihen E und F waren für die HLA-Inhibitionsexperimente vorgesehen. In Reihe G wurden 100μl Medium und später die Chrom markierten Zielzellen für die Ermittlung der spontanen Chromfreisetzung gegeben und in Reihe H wurde schließlich das Detergenz Triton-X zugegeben, um die maximalen Freisetzung des gespeicherten Chroms zu bestimmen. |
Ein Verweis auf die Quelle fehlt. |
|
| [18.] Dml/Fragment 043 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 23:02:30 WiseWoman | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 43, Zeilen: 1ff (komplett) |
Quelle: Regn 2002 Seite(n): 77, 78, 79, Zeilen: 77: letzter Abschnitt; 78: 1ff; 79: 1ff |
|---|---|
| Die Zielzellen wurden geerntet, abzentrifugiert und in 50 μl komplettem RPMI resuspendiert und je nach Kalibrierungsdatum mit 30- 60 μl der verdünnten Natriumchromatlösung (51Cr-Isotop; 0,1 mCi am Kalibrationstag) für 60 Minuten bei 37°C im Inkubator inkubiert. Anschließend wurden die Zellen dreimal mit 5 ml komplettem RPMI gewaschen und schließlich in 1ml komplettem RPMI aufgenommen, gezählt und auf 2x104/ml eingestellt. Die so eingestellten Zielzellen wurden dann zu den wie oben beschriebenen CTLs gegeben, wobei pro CTL zu Zielzell Ratios dreier Ansätze gewählt wurden. Anschließend wurde dieser Testansatz für vier Stunden bei 37°C inkubiert. Das während dieser Zeit an das Medium abgegebene Chrom kann als ein direktes Maß für die Lyse durch die Effektorzellen bzw. für die spontane Radioaktivitätsabgabe einer jeden Zielzelle gewertet werden.
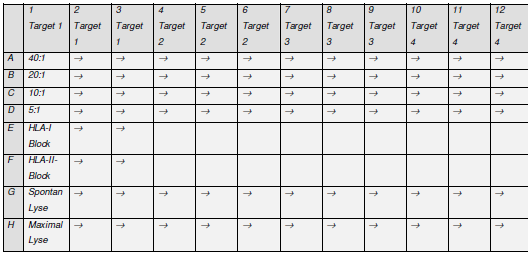
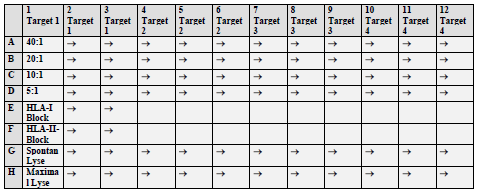
Abbildung 5 gibt einen schematischen Überblick über die experimentelle Anordnung der CTLs und ihren Zielzellen in der Platte
Abbildung 5. Schematische Anordnung einer 96-Well-Spitzbodenplatte wie sie für die Chromfreisetzungsexperimente verwendet wurde. MESSUNG DER SPEZIFISCHEN ZYTOTOXIZITÄT Nach der 4stündigen Inkubationszeit wurden 50 μl des Kulturüberstandes aus jedem Well der Testplatte in die Vertiefungen einer 96 Well Luma-Platte transferiert. Die Lumaplatte ist eine spezielle, für die Messung von Chrom entwickelte Mikrotiterplatte, die mit einer festen Szintillationssubstanz gefüllt ist, [die es dann erlaubt, die resultierende Gamma Strahlung im TOP-Counter zu messen.] |
Die Zielzellen wurden geerntet, abzentrifugiert und in 50 μl komplettem RPMI resuspendiert und je nach Kalibrierungsdatum mit 30- 60 μl der verdünnten Natriumchromatlösung (51Cr-Isotop; 0,1 mCi am Kalibrationstag) für 60 Minuten bei 37°C im Inkubator inkubiert. Anschließend wurden die Zellen dreimal mit 5 ml komplettem RPMI gewaschen und schließlich in 1ml komplettem RPMI aufgenommen, gezählt und auf 2x104/ml eingestellt. Die
[Seite 78] so eingestellten Zielzellen wurden dann zu den wie oben beschriebenen ZTL gegeben, wobei pro ZTL zu Zielzell Ratios Dreier Ansätze gewählt wurden. Anschließend wurde dieser Testansatz für vier Stunden bei 37°C inkubiert. Das während dieser Zeit an das Medium abgegebene Chrom kann als ein direktes Maß für die Lyse durch die Effektorzellen bzw. für die spontane Radioaktivitätsabgabe einer jeden Zielzelle gewertet werden. Abbildung 1 gibt einen schematischen Überblick über die experimentelle Anordnung der ZTL und ihren Zielzellen in der Platte
Abbildung 1: Schematische Anordnung einer 96-Well-Spitzbodenplatte wie sie für die Chromfreisetzungsexperimente verwendet wurde. [Seite 79] Messung der spezifischen Zytotoxizität Nach der 4stündigen Inkubationszeit wurden 50 μl des Kulturüberstandes aus jedem Well der Testplatte in die Vertiefungen einer 96 Well Luma-Platte transferiert. Die Lumaplatte ist eine spezielle, für die Messung von Chrom entwickelte Mikrotiterplatte, die mit einer festen Scintillationssubstanz gefüllt ist, die es dann erlaubt, die resultierende Gamma Strahlung im TOP-Counter zu messen. |
Ein Verweis auf die Quelle fehlt. |
|
| [19.] Dml/Fragment 044 01 - Diskussion Zuletzt bearbeitet: 2018-03-16 21:20:06 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 44, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 79, 80, Zeilen: 79: 3 ff.; 80: 1 ff. |
|---|---|
| [Die Lumaplatte ist eine spezielle, für die Messung von Chrom entwickelte Mikrotiterplatte, die mit einer festen Szintillationssubstanz gefüllt ist,] die es dann erlaubt, die resultierende Gamma Strahlung im TOP-Counter zu messen. Auch hier wird die Radioaktivität für die Dauer einer Minute gemessen (CPM=Counts per minute). Als Spontanwert diente die Chromabgabe jeder Zielzelle ohne die Zugabe von CTLs (Reihe G, Abbildung) und als Maximalwert diente die Chromabgabe, die durch Triton-X- Behandlung der Zielzellen bewirkt wurde (Reihe H, Abbildung). Aus den experimentell erzielten 51Cr- Freisetzungswerten, den spontanen und maximalen 51Cr-Freisetzungswerten läßt sich nach folgender Formel der Prozentsatz der spezifischen Lyse ermitteln:
MHC-INHIBITIONS ASSAY Um zu bestimmen ob die spezifische Zytotoxizität der T-Zellen MHC Klasse-I oder MHC-Klasse-II restringiert ist, wurden die autologen DCs und die autologen LCLs 30 Minuten vor Zugabe der CTLs mit 20 μg/ml der jeweiligen monoklonalen Antikörpern gegen Klasse I und Klasse II inkubiert. Dabei bindet der monoklonale Antikörper W6/32 (DAKO) an monomorphe HLA-ABC- Determinanten und inhibiert dabei deren Erkennung und der monoklonale Antikörper CR3/43 (DAKO) bindet an HLA-DR und verhindert damit HLA-II restringierte Erkennung. IMMUNOPHÄNOTYPISIERUNG DER CTLS MITTELS DIREKTER IMMUNFLUORESZENZ Zur Bestimmung der Oberflächenexpression spezifischer T-Zellmarker wurden die CTLs mit verschiedenen Kombinationen von FITC-oder PE-konjugierten monoklonalen Antikörpern inkubiert und anschließend in der Durchflusszytometrie ausgewertet. Eingesetzt wurden dazu folgende Antikörper: CD-3, CD-8, CD-4, CD-56, CD-16, CD-25, TCRγδ, TCRαβ, CD-2. |
Die Lumaplatte ist eine spezielle, für die Messung von Chrom entwickelte Mikrotiterplatte, die mit einer festen Scintillationssubstanz gefüllt ist, die es dann erlaubt, die resultierende Gamma Strahlung im TOP-Counter zu messen. Auch hier wird die Radioaktivität für die Dauer einer Minute gemessen (CPM=Counts per minute). Als Spontanwert diente die Chromabgabe jeder Zielzelle ohne die Zugabe von ZTL (Reihe G, Abbildung) und als Maximalwert diente die Chromabgabe, die durch Triton-X- Behandlung der Zielzellen bewirkt wurde (Reihe H, Abbildung). Aus den experimentell erzielten 51Cr-Freisetzungswerten, den spontanen und maximalen 51Cr-Freisetzungswerten läßt sich nach folgender Formel der Prozentsatz der spezifischen Lyse ermitteln:
[Seite 80] MHC-Inhibitions- Assay Um zu bestimmen ob die spezifische Zytotoxizität der T-Zellen MHC Klasse-1 oder MHC-Klasse-2 restringiert ist, wurden die autologen AdV-infizierten DCs und die autologen LCLs 30 Minuten vor Zugabe der ZTL mit 20 μg/ml der jeweiligen monoklonalen Antikörpern gegen Klasse I und Klasse II inkubiert. Dabei bindet der monoklonale Antikörper W6/32 (DAKO) an monomorphe HLA-ABC- Determinanten und inhibiert dabei deren Erkennung und der monoklonale Antikörper CR3/43 (DAKO) bindet an HLA-DR, DP und DQ und verhindert damit HLA-II restringierte Erkennung. Immunophänotypisierung der ZTL mittels direkter Immunfluoreszenz Zur Bestimmung der Oberflächenexpression spezifischer T-Zellmarker wurden die ZTL mit verschiedenen Kombinationen von FITC-oder PE-konjugierten monoklonalen Antikörpern inkubiert und anschließend in der FACS-Analyse ausgewertet. Eingesetzt wurden dazu folgende Antikörper: CD3, CD8, CD4, CD56, CD16, CD25, TCRγδ, TCR αβ, CD2. |
Ein Verweis auf die Quelle fehlt. Aus "51Cr" in der Formel für spezifische Lyse in der Quelle wurde "51Cr" - ein typischer Copy & Paste-Fehler. |
|
| [20.] Dml/Fragment 045 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 23:19:14 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 45, Zeilen: 1-12 |
Quelle: Regn 2002 Seite(n): 81, Zeilen: 1 ff. |
|---|---|
| STATISTISCHE AUSWERTUNG DER BEOBACHTETEN ERGEBNISSE
BERECHNUNG DER ARITHMETISCHEN MITTELWERTE Das arithmetische Mittel x ist die Summe aller Beobachtungen geteilt durch die Anzahl dieser Beobachtungen und dient zur repräsentativ-nivellierenden Informationsverdichtung für mehrere bis zahlreiche nicht zu heterogenerDaten [sic].
BERECHNUNG DER STANDARDABWEICHUNG Die Standardabweichung s ist gleich der positiven Quadratwurzel aus dem Mittelwert der quadrierten Abweichungen:
EVALUIERUNG DES SIGNIFIKANZNIVEAUS UND BERECHNUNG DER P-WERTE Das Signifikanzniveau von P< 0,05 wurde festgelegt. Alle Werte, die größer als 0,05 waren wurden als statistisch nicht signifikant gewertet, das heißt die Nullhypothese war abzulehnen. |
Statistische Auswertung der beobachteten Ergebnisse
[...] 1. Berechnung der arithmetischen Mittelwerte Das arithmetische Mittel x ist die Summe aller Beobachtungen geteilt durch die Anzahl dieser Beobachtungen und dient zur repräsentativ-nivellierenden Informationsverdichtung für mehrere bis zahlreiche nicht zu heterogene Daten.
2. Berechnung der Standardabweichung Die Standardabweichung s ist gleich der positiven Quadratwurzel aus dem Mittelwert der quadrierten Abweichungen:
3. Evaluierung des Signifikanzniveaus und Berechnung der P-Werte Das Signifikanzniveau von P< 0,05 wurde festgelegt. Alle Werte, die größer als 0,05 waren wurden als statistisch nicht signifikant gewertet, das heißt die Nullhypothese war abzulehnen. |
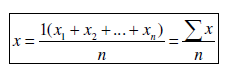
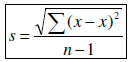
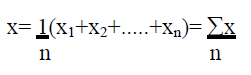
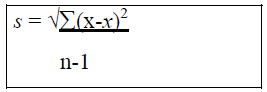
Ein Verweis auf die Quelle fehlt. Die in der Quelle durchaus ungewöhnlich ausgeführten Standardformeln werden genauso in die untersuchte Arbeit übernommen. |
|
| [21.] Dml/Fragment 052 09 - Diskussion Zuletzt bearbeitet: 2018-03-16 21:21:17 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 52, Zeilen: 9-11 |
Quelle: Regn 2002 Seite(n): 85, 86, Zeilen: 85: 10 ff.; 86: Abbildung |
|---|---|
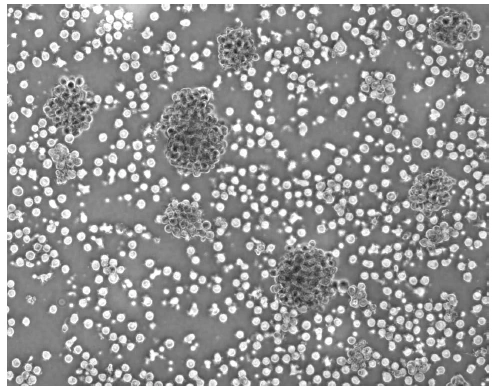
Abbildung 11. Unreife DCs in einer Kulturplatte: Die Zellen aggregieren und bilden sich zu kleineren und größeren Clustern zusammen. Leukozyten wurden aus dem frischen peripheren Blut oder aus „buffy coats“ isoliert und nach der zweistündigen Adhärenzphase in serumfreien Medium, das mit 500U/ml IL-4, 800 U/ml GM-CSF supplementiert war kultiviert. |
Leukozyten wurden aus dem frischen peripheren Blut oder aus „buffy coats“ isoliert und nach der zweistündigen Adhärenzphase in einem komplettem Medium, das mit 1000 U/ml IL-4, 800 U/ml GM-CSF supplementiert war kultiviert.
[Seite 86] Abbildung 1: Unreife DCs in einer Kulturplatte: Die Zellen aggregieren und bilden sich zu kleineren und größeren Clustern zusammen (Pfeile)
|
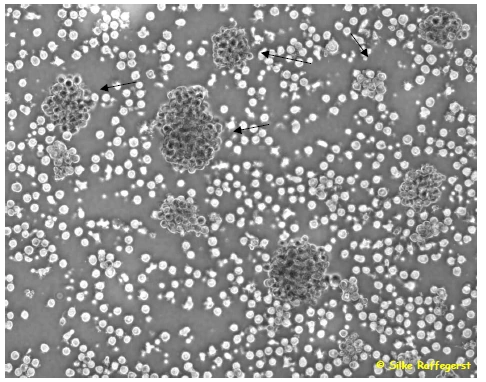
Ein Verweis auf die Quelle fehlt. Man beachte, dass in der in der Quelle wiedergegebenen Version der Abbildung ein Copyright-Vermerk existiert; in der untersuchten Arbeit fehlt dieser. |
|
| [22.] Dml/Fragment 053 01 - Diskussion Zuletzt bearbeitet: 2018-03-16 21:22:13 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 53, Zeilen: 1-11 |
Quelle: Regn 2002 Seite(n): 85, 86, 88, Zeilen: 85: 14 ff.; 86: 1 ff.; 88: Abbildung |
|---|---|
| [Nach acht bis] zehn Tagen konnte bereits lichtmikroskopisch der typische Phänotyp unreifer DCs mit großen granulären Zellkörpern, lobulären Nuclei und feinen zytoplasmatischen Ausläufern detektiert werden. Wie in Abbildung 11. deutlich zu sehen, bilden mehrere DCs kleinere bis größere Cluster auf dem Kulturplattenboden, auf dem sie aber nur leicht anhaften. Man bezeichnet diese Zellen daher als semi-adhärent, da sie keinen konfluenten Zellrasen bilden und man sie durch auf- und abpipettieren mit einer Pasteurpipette leicht von der Platte spülen kann.
Abbildung 12. zeigt den Vergleich zwischen unreifen und reifen dendritischen Zellen anhand eines charakteristischen Beispiels. Die noch unreifen DCs wurden dann nachfolgend mit Hilfe der Durchflußzytometrie und Anfärben mit spezifischen, monoklonalen Antikörpern immunphänotypisch charakterisiert. |
Nach sieben bis acht Tagen konnte bereits lichtmikroskopisch der typische Phänotyp unreifer DCs mit großen granulären Zellkörpern, lobulären Nuclei und feinen zytoplasmatischen Ausläufern detektiert werden. Wie in Abbildung 1 deutlich zu sehen, bilden mehrere DCs kleinere bis größere Cluster auf dem Kulturplattenboden, auf dem sie aber nur leicht anhaften. Man bezeichnet diese Zellen daher als semi-adhärent, da sie keinen konfluenten Zellrasen bilden und man sie durch auf- und abpipettieren mit einer Einweg Pasteurpipette leicht von der Platte spülen kann
[Seite 86] Die noch unreifen DCs wurden dann nachfolgend mit Hilfe der Durchflußzytometrie durch Anfärben mit spezifischen monoklonalen Antikörpern immunphänotypisch charakterisiert. [Seite 88] Abbildung 2: Immunphänotypische Analyse von dendritischen Zellen (DC)
A: Expressionsmuster von 11 Oberflächenmarkern, die am Tag 10 (serumfreie Bedingungen oder Tag 6 serumhaltige Kulturbedingungen) auf unreifen DCs exprimiert werden. Der schwarze Bereich des Histogramms zeigt die Isotypkontrolle spezifisch für jeden verwendeten Antikörper B: Expressionsmuster von CD54, CD11c, CD51, CD1a, CD86, CD40, CD1a, HLA-ABC und HLA-DR nach Zugabe von TNF-α |
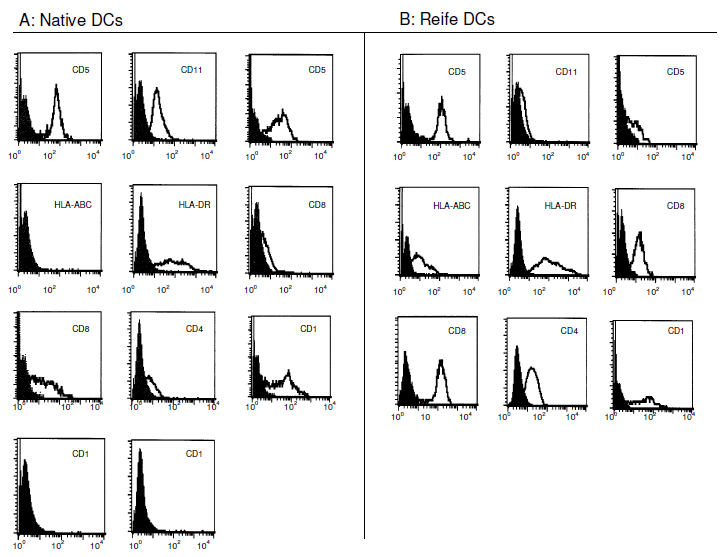
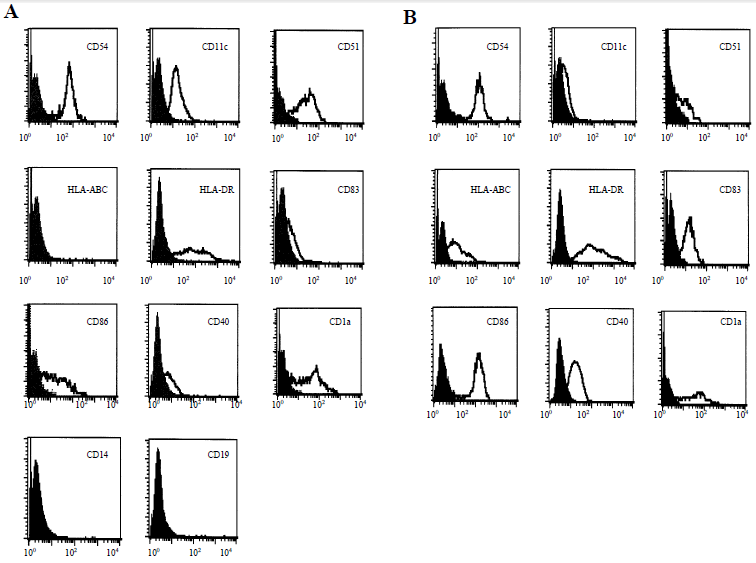
Ein Verweis auf die Quelle fehlt. Die in der Abbildung gezeigten Kurven sind identisch. |
|
| [23.] Dml/Fragment 054 01 - Diskussion Zuletzt bearbeitet: 2018-03-15 17:25:00 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 54, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 86, 87, 89, Zeilen: 86: 3 ff.; 87: 1 ff.; 89: Abbildung |
|---|---|
| [Sie exprimierten weder den Monozyten- und Makrophagenmarker CD14 noch das B-Zell spezifischen Antigen CD19 und] zeigten niedrige bis mittlere Expression der kostimulatorischen und Adhäsionsmoleküle Moleküle CD86, CD40 und CD54 (ICAM-1) sowie geringe bis mittlere Expression der MHC-Moleküle, HLA- ABC und HLA-DR, aber geringe oder gar keine Expression des DC- Markers für reife Zellen CD83. Das Molekül CD1a, [sic] wurde in hoher Konzentration auf den unreifen DC exprimiert, ebenso CD11c und das α5β3-Integrin CD51. Aufgrund dieses Oberflächenprofils wurden die Zellen als unreif charakterisiert.
Abbildung 13. Prozentuale Verteilung der Oberflächenmarker auf reifen und unreifen DCs. Die Werte zeigen die Mittelwerte und Standardabweichungen von acht repräsentativen Experimenten. Die hellen Balken repräsentieren DCs, die in serumfreien Medium für 10-12 Tage in der Anwesenheit von IL-4 und GM-CSF kultiviert wurden. Die dunkelgrauen Balken zeigen die gleichen Zellen, die aber drei Tage länger unter Zusatz von TNF-α zur Reife stimuliert wurden. Die quantitative Auswertung der Zellpopulationen ergab je nach Spender eine Ausbeute von 35 bis 75 %. Die Zugabe von TNF-α (200 U/ml) in das Kulturmedium bewirkte einen Stimulus [sic] der die Zellen zu reifen DCs differenzieren ließ. Diese Zellen veränderten sich phänotypisch durch Vergrößerung des Zellkörpers und das Auswachsen längerer zytoplasmatischer Fortsätze. Außerdem konnte stärkeres Anhaften am Kulturplattenboden bemerkt werden. Wiederholung der Immunphänotypisierung zeigte eine Veränderung des Oberflächenprofiles dahingehend, das [sic] nun die kostimulatorischen Moleküle CD86 [und CD40, die MHC-Moleküle HLA-ABC und HLA-DR, das Adhäsionsmolekül ICAM (CD54), sowie CD83 hochreguliert wurden, während es zu einer deutlichen Verminderung in der Expression der Moleküle CD1a, CD11c sowie dem α5β3-Integrin CD51 kam.] |
Sie waren negativ in der Expression des Monozyten und Makrophagenmarkers CD14 und des B-Zell spezifischen Antigens CD19 und zeigten niedrige bis mittlere Expression der Kostimulatorischen und Adhäsionsmoleküle Moleküle CD86, CD40 und CD54 (ICAM-1) sowie geringe bis mittlere Expression der MHC-Moleküle, HLA- ABC und HLA-DR, aber geringe oder gar keine Expression des DC- Markers für reife Zellen CD83. Das Molekül CD1a, [sic] wurde in hoher Konzentration auf den unreifen DC exprimiert, ebenso CD11c und das α5β3-Integrin CD51.
Aufgrund dieses Oberflächenprofils wurden die Zellen als unreif charakterisiert. Die quantitative Auswertung der Zellpopulationen ergab je nach Spender eine Ausbeute von 25 bis 35 %. [Seite 87] Die Zugabe von TNF-α (200 U/ml) oder IL-1β (20 ng/ml) in das Kulturmedium bewirkte einen Stimulus [sic] der die Zellen zu reifen DCs differenzieren ließ. Diese Zellen veränderten sich phänotypisch durch Vergrößerung des Zellkörpers und das Auswachsen längerer zytoplasmatischer Fortsätze. Außerdem konnte stärkeres Anhaften am Kulturplattenboden bemerkt werden. Wiederholung der Immunphänotypisierung zeigte eine Veränderung des Oberflächenprofiles dahingehend, das [sic] nun die kostimulatorischen Moleküle CD86 und CD40, die MHC-Moleküle HLA-ABC und HLA-DR, das Adhäsionsmolekül ICAM (CD54), sowie CD83 hochreguliert wurden, während es zu einer deutlichen Verminderung in der Expression der Moleküle CD1a, CD11c sowie dem α5β3-Integrin CD51 kam. [Seite 89]
Abbildung 3: Prozentuale Verteilung der Oberflächenmarker auf reifen und unreifen DCs. |
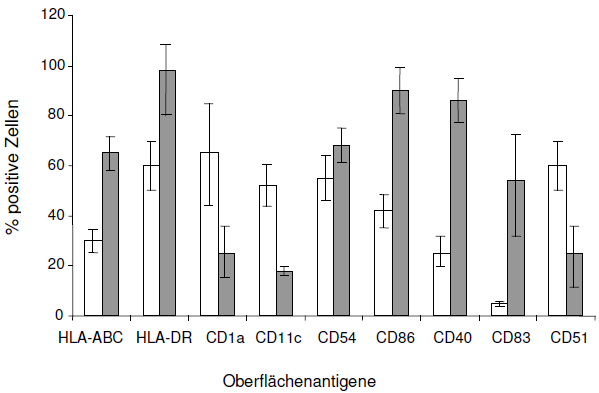
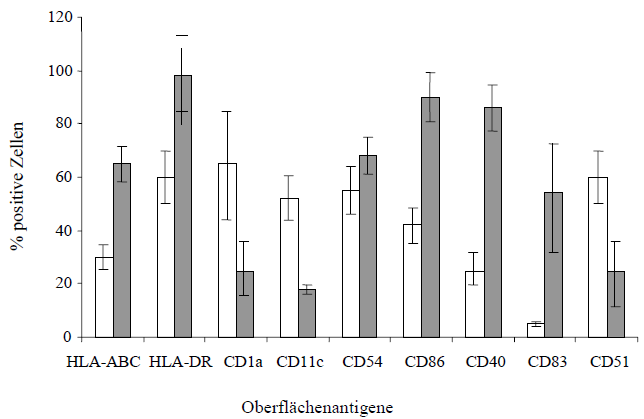
Ein Verweis auf die Quelle fehlt. Die in der Abbildung veranschaulichten Daten sind identisch inklusive Fehlerbalken. Dies ist verwunderlich, da diesen Daten bei der Quelle sechs repräsentative Experimente zu Grunde liegen, bei der untersuchten Arbeit aber acht. |
|
| [24.] Dml/Fragment 055 01 - Diskussion Zuletzt bearbeitet: 2018-03-15 21:39:59 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 55, Zeilen: 1 ff. (komplett) |
Quelle: Regn 2002 Seite(n): 87, 91, Zeilen: 87: 5 ff.; 91: 1 ff. |
|---|---|
| [Wiederholung der Immunphänotypisierung zeigte eine Veränderung des Oberflächenprofiles dahingehend, das [sic] nun die kostimulatorischen Moleküle CD86] und CD40, die MHC-Moleküle HLA-ABC und HLA-DR, das Adhäsionsmolekül ICAM (CD54), sowie CD83 hochreguliert wurden, während es zu einer deutlichen Verminderung in der Expression der Moleküle CD1a, CD11c sowie dem α5β3-Integrin CD51 kam.
TESTSYSTEME ZUR ÜBERPRÜFUNG DER FUNKTIONALITÄT VON DCS UNTERSUCHUNG DER AUFNAHMEKAPAZITÄT VON FITC-DEXTRAN ÜBER DEN MANNOSEREZEPTOR Nach Beurteilung des immunologischen Profils und der quantitativen Ausbeute der DCs wurde die Funktionalität dieser Zellen in verschiedenen Testsystemen überprüft. Ein wichtiges Experiment um die natürliche Kapazität der DCs zu testen ist Makromoleküle über den Mannoserezeptor aufzunehmen, wurden unreife DCs über verschiedene Zeiträume mit FITC markiertem Dextran inkubiert. Anschließend wurde dann die Aufnahme des Dextrans anhand der MFI (mittleren Fluoreszenzintensität) in der Durchflußzytometrie ermittelt. Dabei wurde der Anstiegsfaktor der MFI ausgehend von der Eigenfluoreszens der DCs berechnet. DCs sind große granuläre Zellen von unregelmäßiger Gestalt und unterscheiden sich daher oft in der Ausprägung ihrer Eigenfluoreszens, so daß sich zwischen den einzelnen DC-Populationen unterschiedlicher Spender, auch unterschiedliche Werte für die Eigenfluoreszenz und damit auch in Bezug auf die tatsächlich gemessenen MFI Werte ergeben. Berechnet man aber den Anstieg der MFI gegenüber dieser Eigenfluoreszenz, lassen sich die Werte vieler Experimente miteinander vergleichen. Ein typisches Beispiel für die Aufnahmekapazität unreifer und reifer DCs ist in Abbildung 5 dargestellt. Dabei hat sich gezeigt, dass die MFI im Vergleich zur unbehandelten Kontrolle bei 30 Minuten ansteigt, einen Sättigungsgrad erreicht, bis schließlich kein Dextran mehr aufgenommen werden kann. Der Anstieg der mittleren Fluoreszenzintensität wurde gegenüber der Eigenfluoreszenz korrigiert. Bei unreifen DCs lag die maximale Aufnahmekapazität des FITC-Dextrans bei 120 Minuten und zeigte eine signifikante Abnahme der Aufnahmekapazität bei [180 Minuten.] |
Wiederholung der Immunphänotypisierung zeigte eine Veränderung des Oberflächenprofiles dahingehend, das [sic] nun die kostimulatorischen Moleküle CD86 und CD40, die MHC-Moleküle HLA-ABC und HLA-DR, das Adhäsionsmolekül ICAM (CD54), sowie CD83 hochreguliert wurden, während es zu einer deutlichen Verminderung in der Expression der Moleküle CD1a, CD11c sowie dem α5β3-Integrin CD51 kam.
[Seite 91] Testsysteme zur Überprüfung der Funktionalität von DCs a) Untersuchung der Aufnahmekapazität von FITC-Dextran über den Mannoserezeptor Nach Beurteilung des immunologischen Profils und der quantitativen Ausbeute der DCs wurde die Funktionalität dieser Zellen in verschiedenen Testsystemen überprüft. Ein wichtiges Experiment um die natürliche Kapazität der DCs zu testen Makromoleküle über den Mannoserezeptor aufzunehmen, wurden unreife DCs unter beiden Kulturbedingungen über verschiedene Zeiträume mit FITC markiertem Dextran inkubiert. Anschließend wurde dann die Aufnahme des Dextrans anhand der MFI (mittleren Fluoreszenzintensität) in der Durchflußzytometrie ermittelt. Dabei wurde der Anstiegsfaktor der MFI ausgehend von der Eigenfluoreszens der DCs berechnet. DCs sind große granuläre Zellen von unregelmäßiger Gestalt und unterscheiden sich daher oft in der Ausprägung ihrer Eigenfluoreszens, sodaß sich zwischen den einzelnen DC-Populationen unterschiedlicher Spender auch unterschiedliche Werte für die Eigenfluoreszenz und damit auch in Bezug auf die tatsächlich gemessenen MFI Werte ergeben. Berechnet man aber den Anstieg der MFI gegenüber dieser Eigenfluoreszenz, lassen sich die Werte vieler Experimente miteinander vergleichen. Ein typisches Beispiel für die Aufnahmekapazität unreifer und reifer DCs ist in Abbildung 5 dargestellt. Dabei hat sich gezeigt, dass die MFI im Vergleich zur unbehandelten Kontrolle bei beiden Gruppen bei 30 Minuten ansteigt, einen Sättigungsgrad erreicht bis schließlich kein Dextran mehr aufgenommen werden kann. Der Anstieg der mittleren Fluoreszenzintensität wurde gegenüber der Eigenfluoreszenz korrigiert. In Unabhängigkeit von den Kulturbedingungen der unreifen DCs lag die maximale Aufnahmekapazität des FITC-Dextrans bei 120 Minuten und zeigte eine signifikante Abnahme der Aufnahmekapazität bei 180 Minuten. |
Ein Verweis auf die Quelle fehlt. |
|
| [25.] Dml/Fragment 056 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 22:56:08 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 56, Zeilen: 1-10 |
Quelle: Regn 2002 Seite(n): 91, 92, Zeilen: 91: 23 ff.; 92: 1 ff. |
|---|---|
| Die gleichen Experimente wurden mit reifen DCs durchgeführt und wie in der Abbildung dargestellt, verlief die Kurve, aufgrund der eingeschränkten Aufnahmekapazität reifer DCs, deutlich flacher als bei den unreifen Zellen und zeigte auch keinen signifikanten Anstieg oder Abstieg der Aufnahmekapazität.
Abbildung 14. Aufnahme durch FITC-Dextran durch unreife DCs (dicke Linie) im Vergleich zu reifen DCs (dünne Linie) Unreife dendritische Zellen verfügen über eine höhere Kapazität über den Mannoserezeptor FITC-konfugiertes Dextran aufzunehmen als reife DCs. Die durchgezogenen Linie zeigt über einen Zeitraum von 30 bis 180 Minuten die Fähigkeit unreifer DCs das Makromolekül FITC-Dextran aufzunehmen. Die gestrichelte Linie zeigt reife dendritische Zellen und deren eingeschränkte Aufnahmekapazität gegenüber FITC-Dextran. |
Die gleichen Experimente wurden mit reifen DCs durchgeführt und wie in der Abbildung dargestellt verlief die Kurve, aufgrund der eingeschränkten Aufnahmekapazität reifer DCs deutlich flacher als bei den unreifen Zellen und zeigte auch keinen signifikanten Anstieg oder Abstieg der Aufnahmekapazität.
[Seite 92] Unreife Dendritische Zellen verfügen über eine höhere Kapazität über den Mannoserezeptor FITC-konfugiertes Dextran aufzunehmen als reife DCs. Die blaue Linie zeigt über einen Zeitraum von 30 bis 180 Minuten die Fähigkeit unreifer DCs das Makromolekül FITC-Dextran aufzunehmen. Die graue Linie zeigt reife Dendritische Zellen und deren eingeschränkte Aufnahmekapazität gegenüber FITC-Dextran.
Abbildung 5: Aufnahme durch FITC-Dextran durch unreife DCs im Vergleich zu reifen DCs |
Ein Verweis auf die Quelle fehlt. |
|
| [26.] Dml/Fragment 057 08 - Diskussion Zuletzt bearbeitet: 2018-03-12 17:08:09 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 57, Zeilen: 8-31 |
Quelle: Regn 2002 Seite(n): 110, Zeilen: 11 ff. |
|---|---|
| Um die immunstimulatorische Kapazität von mit Tumor-RNA-transduzierten DCs zu testen wurden gemischte Lymphozyten Reaktions Experimente (MLR= mixed lymphocyte reaction) durchgeführt. Ursprünglich, diente diese Methode zum Nachweis von Gewebeunverträglichkeit. In dem klassischen Test werden gleiche Mengen von bestrahlten Stimulator-Lymphozyten mit unbestrahlten Responder- Lymphozyten inkubiert um eine aussagekräftige Reaktion zu erhalten. Wenn man aber DCs als Stimulatoren verwendet, reichen weniger Zellen aus um die T-Zellen zur Proliferation anzuregen.
Für den allogenen Ansatz wurde aus dem peripheren Blut von HLA-inkompatiblen gesunden Spendern T-Zellen über ein negatives Zellseparationsverfahren isoliert. Dazu wurden die PBMNCs mit einer Mischung spezifischer Antikörper (CD11b, CD56, CD36, CD19), die gegen Nicht-T-Zellen gerichtet waren markiert. An diese Antikörper sind magnetische „Beads“ gekoppelt, so dass nach Auftragen der Zellsuspension auf eine mit Stahlkügelchen gefüllte Säule, die an einen Magneten installiert wurde, alle markierten Zellen in der Säule hängen blieben, während die unmarkierte negative Zellfraktion durch die Säule lief und für die weitere experimentelle Vorgehensweise gesammelt wurde. Diese von der Firma Miltenyi entwickelte Methode garantiert eine 98 %ige Reinheit an CD3-positiven T-Zellen und dies konnte auch durch Anfärben mit monoklonalen Antikörpern, die gegen CD3 gerichtet waren, in der Durchflußzytometrie bestätigt werden. Schließlich wurden variierende Zahlen (10000, 5000, 2000) von bestrahlten DCs, die aus dem Blut von SCCHN Patienten gewonnen wurden mit 1x105 dieser hochreinen T-Zellen kokultiviert. Dabei ergaben sich Stimulator zu Responder Ratios von 10:1, 20:1 und 50:1. |
Um die immunstimulatorische Kapazität der AdV-infizierten DCs zu testen wurden gemischte Lymphozyten Reaktions Experimente (MLR= mixed lymphozyte [sic] reaction) durchgeführt. Wie bereits bei der Beschreibung der in serumhaltigem Medium kultivierten DCs erwähnt, diente diese Methode ursprünglich zum Nachweis von Gewebeunverträglichkeit. In dem klassischen Test werden gleiche Mengen von bestrahlten Stimulator-Lymphozyten mit unbestrahlten Responder- Lymphozyten inkubiert um eine aussagekräftige Reaktion zu erhalten. Wenn man aber DCs als Stimulatoren verwendet, reichen weniger Zellen aus um die T-Zellen zur Proliferation anzuregen.
Aus dem peripheren Blut von AdV-seropositiven, HLA-inkompatiblen Spendern wurden T-Zellen über ein negatives Zellseparationsverfahren isoliert. Dazu wurden die PBMNCs mit einer Mischung spezifischer Antikörper (CD11b, CD56, CD36, CD19), die gegen Nicht-T-Zellen gerichtet waren markiert. An diese Antikörper sind magnetische Beads gekoppelt, so dass nach Auftragen der Zellsuspension auf eine mit Stahlkügelchen gefüllte Säule, die an einen Magneten installiert wurde alle markierten Zellen in der Säule hängen blieben, während die unmarkierte negative Zellfraktion durch die Säule lief und für die weitere experimentelle Vorgehensweise gesammelt wurde. Diese von der Firma Miltenyi entwickelte Methode garantiert eine 98 %ige Reinheit an CD3-positiven T-Zellen und dies konnte auch durch Anfärben mit monoklonalen Antikörpern, die gegen CD3 gerichtet waren, in der Durchflußzytometrie bestätigt werden. Schließlich wurden variierende Zahlen (10000, 2500, 1000) von bestrahlten infizierten und uninfizierten DCs mit 1x105 dieser hochreinen T-Zellen kokultiviert. Dabei ergaben sich Stimulator zu Responder Ratios von 10:1, 40:1 und 100:1. |
Ein Verweis auf die Quelle fehlt. |
|
| [27.] Dml/Fragment 058 01 - Diskussion Zuletzt bearbeitet: 2018-03-10 19:01:25 Schumann | Dml, Fragment, Gesichtet, KomplettPlagiat, Regn 2002, SMWFragment, Schutzlevel sysop |
|
|
|
| Untersuchte Arbeit: Seite: 58, Zeilen: 1-10 |
Quelle: Regn 2002 Seite(n): 111, Zeilen: 1 ff. |
|---|---|
| Obwohl DCs sich nicht mehr teilende Zellen sind, wurden sie deshalb bestrahlt, weil es sich bei den DC-Kulturen niemals um reine Kulturen handelte und sie immer über einen gewissen Anteil an Lymphozyten verfügten, deren Teilung in der MLR nicht erwünscht waren. Nach sechs Tagen wurde der MLR 3H-markiertes Thymidin für einen Zeitraum von 18 Stunden zugesetzt. Das radioaktiv markierte Thymidin baut sich nur in die DNA von teilenden Zellen ein. Die emittierende β-Strahlung kann dann nach Ernten der Zellen als cpm (Scintillationsmeßwert pro Minute) gemessen werden und stellt ein Maß für die Proliferationskapazität der Zellen dar. Die Ergebnisse dieser Experimente (n=6) wurden in Abbildung 15. zusammengefasst. | Obwohl DCs sich nicht mehr teilende Zellen sind, wurden sie deshalb bestrahlt, weil es sich bei den DC-Kulturen niemals um reine Kulturen handelte und sie immer über einen gewissen Anteil an Lymphozyten verfügten, deren Teilung in der MLR nicht erwünscht waren. Nach sechs Tagen wurde der MLR 3H-markiertes Thymidin für einen Zeitraum von 18 Stunden zugesetzt. Das radioaktiv markierte Thymidin baut sich nur in die DNA von teilenden Zellen ein. Die emittierende β-Strahlung kann dann nach Ernten der Zellen als cpm (Scintillationsmeßwert pro Minute) gemessen werden und stellt ein Maß für die Proliferationskapazität der Zellen dar. Die Ergebnisse dieser Experimente (n=6) wurden in Abbildung 18 zusammengefaßt. |
Ein Verweis auf die Quelle fehlt. Die folgenden Abbildungen sind nicht identisch. |
|
| [28.] Dml/Fragment 064 01 - Diskussion Zuletzt bearbeitet: 2018-03-15 22:27:51 WiseWoman | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 64, Zeilen: 1-27 |
Quelle: Regn 2002 Seite(n): 115, 116, Zeilen: 115: 9ff; 116: 3ff |
|---|---|
| Aus frischem autologen Spenderblut isolierte PBL (ggf kryokonserviert unmittelbar vor dem Ansetzen der Kokultur aufgetaute autologe PBL) dienten als Responderzellen. Dabei wurden die Stimulatorzellen diesmal nicht bestrahlt, da die Lymphozyten innnerhalb der DC-Population im autologen System keinen Einfluß auf die Stimulationskapazität der DCs hatten. Das Verhältnis der Stimulator zu Responderzellen betrug in der der Startkultur 5x104 AdV+DCs zu 2x106 PBMNCs. Nach zehn Tagen wurde die sich entwickelnde ZTL-Kultur zum ersten Mal mit autologen AdV+DCs in einer Ratio von 4:1 (ZTL:DC) und danach noch einmal mit autologen LCLs (4:1) restimuliert [sic] wobei am Tag 17 niedrigdosiertes IL-2 (20 U/ml) dem Kulturmedium zugefügt wurde. Der Zeitpunkt und die Menge des zugefügten IL-2 waren entscheidend um das Auswachsen unspezifischer Effektorzellen zu verhindern, die sich zu Beginn in großen Mengen innerhalb der gesamten T-Zellpopulation in der Kultur aufhalten. Zu frühe Gabe von IL-2, dem für die Aktivierung und Proliferation von T-Zellen wichtigsten Wachstumsfaktor, würde selbst die Proliferation solcher Zellen bewirken, die nur die niederaffine Form des IL-2-Rezeptors exprimieren. Diese Zellen könnten dann das Expandieren der spezifischen T-Zellklone beeinträchtigen. Zur Bestimmung der spezifische Zytotoxizität diente der Standard Chromfreisetzungs Testes [sic] (chrom 51-release Assay) als Methode der Wahl (Abbildung 17)
In diesem Test wurden die Effektorzellen (ZTL) mit variierenden Zellzahlverhältnis von 51Chrommarkierten Zielzellen (Targets) für vier Stunden inkubiert. Das Verhältnis der Effektoren zu den Targets reichte dabei von 5:1, 10:1, 20:1 bis 40:1. Dendritische Zellen wurden auch als Zielzellen verwendet. Die Zielzellen, die getestet wurden [sic] waren:, [sic] Tumorzellinie GHD, HLA-mismatched DCs, autologe und allogene LCLs, sowie die T-Zellinie HSB-2, die aufgrund einer Mutation in den MHC-I-Genen von NK-Zellen erkannt und lysiert wird. |
Aus frischem autologen Spenderblut isolierte PBL (falls kryokonserviert unmittelbar vor dem Ansetzen der Kokultur aufgetaute autologe PBL) dienten als Responderzellen. Dabei wurden die Stimulatorzellen diesmal nicht bestrahlt, da die Lymphozyten innnerhalb der DC-Population im autologen System keinen Einfluß auf die Stimulationskapazität der DCs hatten. Das Verhältnis der Stimulator zu Responderzellen betrug in der der Startkultur 5x104 AdV+DCs zu 2x106 PBL. Nach zehn Tagen wurde die sich entwickelnde ZTL-Kultur zum ersten Mal mit autologen AdV+DCs in einer Ratio von 4:1 (ZTL:DC) und danach noch wöchentlich mit autologen LCLs (4:1) restimuliert [sic] wobei am Tag 17 niedrigdosiertes IL-2 (20 U/ml) dem Kulturmedium zugefügt wurde. Der Zeitpunkt und die Menge des zugefügten IL-2 waren entscheidend um das Auswachsen unspezifischer Effektorzellen zu verhindern, die sich zu Beginn in großen Mengen innerhalb der gesamten T-Zellpopulation in der Kultur aufhalten. Zu frühe Gabe von IL-2, dem für die Aktivierung und Proliferation von T-Zellen wichtigsten Wachstumsfaktor, würde selbst die Proliferation solcher Zellen bewirken, die nur die niederaffine Form des IL-2-Rezeptors exprimieren. Diese Zellen könnten dann das Expandieren der spezifischen T-Zellklone beeinträchtigen. [...]
[Seite 116] [...] In diesem Test wurden die Effektorzellen (ZTL) mit variierenden Ratios von 51Chrommarkierten Zielzellen (Targets) für vier Stunden inkubiert. Das Verhältnis der Effektoren zu den Targets reichte dabei von 5:1, 10:1, 20:1 bis 40:1. Dendritische Zellen wurden in allen Zytotoxizitätstests auch als Zielzellen verwendet. Die Reihe von Zielzellen, an denen die ZTL-Linien getestet wurden [sic] waren: AdV-infizierte autologe DCs, uninfizierte autologe DCs, HLA-mismatched uninfizierte und AdV-infizierte DCs, autologe und allogene LCLs, sowie die T-Zellinie HSB-2, die aufgrund einer Mutation in den MHC-I-Genen von NK-Zellen erkannt und lysiert wird. |
Ein Verweis auf die Quelle fehlt. |
|
| [29.] Dml/Fragment 075 06 - Diskussion Zuletzt bearbeitet: 2018-03-15 22:20:29 WiseWoman | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 75, Zeilen: 6-27 |
Quelle: Regn 2002 Seite(n): 17, 147, 148, Zeilen: 17: 7ff; 147: letzter Abschnitt; 148: 1ff |
|---|---|
| DCs sind spezialisierte antigenpräsentierende Zellen und verfügen über die Kapazität fremdes Antigen in großen Mengen aufzunehmen und zu verarbeiten. Darüber hinaus sind sie in der Lage diese Antigene naiven oder ruhenden T-Zellen in vitro und in vivo zu präsentieren. Diese APCs exprimieren große Mengen an MHC- Klasse I und II Molekülen und Adhäsionsmolekülen, sowie eine beträchtliche Anzahl der kostimulatorischen Moleküle CD80, CD86 and CD40, welche essentiell für die T-Zellaktivierung sind.
In aktuellen Studien konnte gezeigt werden, daß DCs, die mit spezifischen (Bell et al 1999, Banchereau et al. 1998). [sic!] Zudem haben In [sic] vitro Studien deutlich demonstrieren können, dass DCs, die entweder genetisch modifiziert wurden, um spezifische Antigene zu exprimieren (Brossart et al. 1997, Song et al. 1997), mit Peptiden oder mit Tumorantigenen (Ashley et al. 1997, Boczkowski et al. 1998, Nair et al. 1998) beladen oder mit tumorspezifischer RNA (Nair et al. 1998, Su et al. 2001) gepulst wurden, in der Lage waren hoch effizient antigenspezifische CTL-Antworten hervorzurufen. Diese Erkenntnisse wurden gestützt durch experimentelle Ergebnisse von Studien an Tiermodellen (Zitvogel et al. 1996), sowie erste klinische Studien (Nestlé et al. 1998, Murphy et al. 1996, Tjoa et al. 1998, Hsu et al. 1996) die die Hypothese unterstützen, dass Individuen, die mit Tumorpeptid-beladenen DCs vakziniert wurden, eine tumorspezifische humorale und zelluläre Antwort hervorrufen konnten. Dies führte zum Rückgang der Tumorlast und dem Aufbau einer schützenden Immunität gegenüber neuem Tumorwachstum in vivo. |
DCs sind die einzigen APCs, die in der Lage sind primäre Immunantworten hervorzurufen und sogar naive CD8+ zytotoxische T-Zellen zu stimulieren (Bell et al 1999, Banchereau et al. 1998). In vitro Studien haben deutlich demonstrieren können, daß DCs, die entweder genetisch modifiziert wurden um spezifische Antigene zu exprimieren (Brossart et al. 1997, Song et al. 1997), mit Peptiden oder mit Tumorantigenen (Ashley et al. 1997, Boczkowski et al. 1998, Nair et al. 1998) beladen oder mit tumorspezifischer RNA (Nair et al. 1998, Su et al. 2001) gepulsed wurden, in der Lage waren hoch effizient antigenspezifische ZTL-Antworten hervorzurufen.
Experimentelle Ergebnisse von Studien an Tiermodellen (Zitvogel et al. 1996), sowie erste klinische Studien (Nestlé et al. 1998, Murphy et al. 1996, Tjoa et al. 1998, Hsu et al. 1996) unterstützen die Hypothese, daß Individuen, die mit Tumorpeptid-beladenen DCs vacciniert wurden, eine tumorspezifische humorale und zelluläre Antwort hervorrufen konnten. Dies führte zum Rückgang der Tumorlast und dem Aufbau einer schützenden Immunität gegenüber neuem Tumorwachstum in vivo. [Seite 147] DCs sind spezialisierte antigenpräsentierende Zellen und verfügen über die Kapazität fremdes Antigen in großen Mengen aufzunehmen und zu verarbeiten. Darüber hinaus sind sie in der Lage diese Antigene naiven oder ruhenden T-Zellen in vitro und in vivo zu präsentieren. Diese APCs exprimieren große Mengen an MHC- Klasse I und II Molekülen und Adhäsionsmolekülen, sowie eine beträchtliche Anzahl der kostimulatorischen Moleküle CD80, CD86 and CD40, welche essentiell für die T-Zellaktivierung sind. [Seite 148] In aktuellen Studien konnte gezeigt werden, daß DCs, die mit spezifischen Antigenen in Form von Proteinen, Peptiden oder RNA beladen wurden, sowohl CD8+ als auch CD4+ Tumor- und Virusspezifische ZTL in vitro und in vivo stimulieren konnten. |
Ein Verweis auf die Quelle fehlt. Ein in der Quelle vollständiger Satz ist in der untersuchten Arbeit abgeschnitten, siehe auch Dml/Fragment 081 02. |
|
| [30.] Dml/Fragment 081 02 - Diskussion Zuletzt bearbeitet: 2018-03-11 18:34:05 Schumann | Dml, Fragment, Gesichtet, Regn 2002, SMWFragment, Schutzlevel sysop, Verschleierung |
|
|
|
| Untersuchte Arbeit: Seite: 81, Zeilen: 2-4 |
Quelle: Regn 2002 Seite(n): 148, Zeilen: 1-3 |
|---|---|
| Hintergrund: Dendritische Zellen (=DCs) [sic] die mit spezifischen Antigenen in Form von Proteinen, Peptiden oder RNA beladen werden [sic] können sowohl CD8+ als auch CD4+ Tumor- und Virus-spezifische ZTL in vitro und in vivo stimulieren. | In aktuellen Studien konnte gezeigt werden, daß DCs, die mit spezifischen Antigenen in Form von Proteinen, Peptiden oder RNA beladen wurden, sowohl CD8+ als auch CD4+ Tumor- und Virusspezifische ZTL in vitro und in vivo stimulieren konnten. |
Kein Hinweis auf eine Übernahme. |
|