Angaben zur Quelle [Bearbeiten]
| Autor | Daniel Mauermann |
| Titel | Prävalenz der 3020insC Mutation des NOD2/CARD15-Gens bei Patienten mit chronischer Parodontitis |
| Ort | München |
| Jahr | 2004 |
| Anmerkung | Dissertation zum Erwerb des Doktorgrades der Zahnheilkunde an der Medizinischen Fakultät der Ludwig-Maximilians-Universität zu München |
| URL | http://edoc.ub.uni-muenchen.de/1996/1/Mauermann_Daniel.pdf |
Literaturverz. |
nein |
| Fußnoten | nein |
| Fragmente | 6 |
| [1.] Ves/Fragment 084 03 - Diskussion Zuletzt bearbeitet: 2014-06-21 09:05:35 Hindemith | Fragment, Gesichtet, Mauermann 2004, SMWFragment, Schutzlevel sysop, Verschleierung, Ves |
|
|
|
| Untersuchte Arbeit: Seite: 84, Zeilen: 3-30 |
Quelle: Mauermann 2004 Seite(n): 39, 40, Zeilen: 39: 1 ff.; 40: 1 f. |
|---|---|
| 3.2.5. Polymerasekettenreaktion (PCR)
Mit Hilfe der PCR kann eine bestimmte Zielsequenz einer komplexen genomischen DNA oder bei der RT-PCR eine bestimmte cDNA selektiv exponentiell amplifiziert werden. Die Methode besitzt eine hohe Sensitivität, es reichen wenige Kopien der Zielsequenz als Vorlage aus. Zusätzlich weist sie eine hohe Spezifität auf, eine selektive Amplifizierung einer einzelnen Sequenz unter Millionen verschiedener Sequenzen ist möglich. Dazu müssen sich zwei zu je einem der beiden Stränge des DNA-Doppelstrangs komplementär und mit ihren 3’-Enden aufeinander zu orientieren, etwa 15 – 25 Basen lange Oligonukleotide als Primer an die zuvor denaturierte DNA binden. Dadurch kann eine hitzestabile DNA-Polymerase den zwischen den Primern gelegenen Abschnitt amplifizieren. Zur Minimierung des Auftretens unspezifischer Amplifikate kann eine Taq-DNA-Polymerase verwendet werden, welche hitzelabile Schutzgruppen trägt und erst durch eine Erhitzung auf 95 °C aktiviert wird. Dieser zyklische Prozess aus Denaturierung, Primeranlagerung und Extension wurde so oft wiederholt, bis die Zielsequenz zum Nachweis ausreichend vervielfältigt war. Anfangs erfolgte eine einmalige Anfangsdenaturierung von 5 – 15 Minuten je nach verwendeter Taq- DNA-Polymerase. Danach folgt die beschriebene Amplifikation für 30- 40 Zyklen, bestehend aus Denaturierung bei 94 °C für 30 Sekunden, Primeranlagerung bei der für das betreffende Primerpaar optimalen Temperatur für ebenfalls 30 Sekunden, sowie die Extension bei 72 °C für eine von der Länge des Amplifikats abhängigen Dauer, mindestens jedoch 30 Sekunden. Der letzte Schritt ist die Endextension bei 72 °C für 10 Minuten, bei der alle noch nicht komplett amplifizierten PCR-Produkte vervollständigt werden sollen. Der PCR-Ansatz setzt sich zusammen aus einem PCR-Puffer, einem Mix aus den vier Desoxinukliotidtriphospaten (dATP, dGTP, dCTP, dTTP), den beiden für die Zielsequenz spezifischen Oligonukleotid- Primern, Magnesiumchloridlösung zur Einstellung der durch Austestung ermittelten optimalen Mg2+-Konzentration, der Taq-DNA-Polymerase, destilliertem Wasser zur Einstellung des Gesamtvolumens, sowie der als Matrize dienenden Ausgangs-DNA. Bei jedem PCR-Ansatz wurde eine Negativkontrolle mit Wasser anstatt der Ausgangs-DNA mitgeführt. |
6.2.5 Polymerasekettenreaktion (PCR)
Mit Hilfe der PCR kann eine bestimmte Zielsequenz einer komplexen genomischen DNA oder bei der RT-PCR eine bestimmte cDNA selektiv exponentiell amplifiziert werden. Die Methode besitzt eine hohe Sensitivität, es reichen wenige Kopien der Zielsequenz als Vorlage aus. Zusätzlich weist sie eine hohe Spezifität auf, eine selektive Amplifizierung einer einzelnen Sequenz unter Millionen verschiedener Sequenzen ist möglich. Dazu müssen sich zwei zu je einem der beiden Stränge des DNA-Doppelstrangs komplementäre und mit ihren 3’-Enden aufeinander zu orientierte, etwa 15–25 Basen lange Oligonukleotide als Primer an die zuvor denaturierte DNA binden. Dadurch kann eine hitzestabile DNA-Polymerase den zwischen den Primern gelegenen Abschnitt amplifizieren. Zur Minimierung des Auftretens unspezifischer Amplifikate kann eine Heißstart Taq-DNA-Polymerase verwendet werden, welche hitzelabile Schutzgruppen trägt und erst durch eine Erhitzung auf 95 °C aktiviert wird. Dieser zyklische Prozess aus Denaturierung, Primeranlagerung („Primer-Annealing“) und Extension wurde so oft wiederholt, bis die Zielsequenz zum Nachweis ausreichend vervielfältigt war. Anfangs erfolgte eine einmalige Anfangsdenaturierung von 5-15 Minuten je nach verwendeter Taq-DNA-Polymerase. Danach folgt die beschriebene Amplifikation 30-40 Zyklen bestehend aus Denaturierung bei 94 °C für 30 Sekunden, Primeranlagerung bei der für das betreffende Primerpaar optimalen Temperatur für ebenfalls 30 Sekunden sowie die Extension bei 72 °C für eine von der Länge des Amplifikats abhängigen Dauer, mindestens jedoch 30 Sekunden. Der letzte Schritt ist die Endextension bei 72 °C für 10 Minuten, bei der alle noch nicht komplett amplifizierten PCR-Produkten vervollständigt werden sollen. Der PCR-Ansatz setzt sich zusammen aus einem PCR-Puffer, einem Mix aus den vier Deoxinukleotidtriphosphaten (dATP, dGTP, dCTP, dTTP), den beiden für die Zielsequenz spezifischen Oligonukleotid-Primern, Magnesiumchloridlösung zur Einstellung der durch Austestung ermittelten optimalen Mg2+-Konzentration, der Taq-DNA-Polymerase, destilliertem Wasser zur Einstellung des Gesamtvolumens sowie [Seite 40] der als Matrize dienenden Ausgangs-DNA. Bei jedem PCR-Ansatz wurde eine Negativkontrolle mit Wasser anstatt der Ausgangs-DNA mitgeführt. |
Ein Verweis auf die Quelle fehlt. |
|
| [2.] Ves/Fragment 085 01 - Diskussion Zuletzt bearbeitet: 2014-07-01 14:49:13 Schumann | Fragment, Gesichtet, KomplettPlagiat, Mauermann 2004, SMWFragment, Schutzlevel sysop, Ves |
|
|
|
| Untersuchte Arbeit: Seite: 85, Zeilen: 1 ff. (komplett) |
Quelle: Mauermann 2004 Seite(n): 40, 41, Zeilen: 40: 3 ff.; 41: 1 ff. |
|---|---|
| 3.2.6. Agarosegelelektrophorese
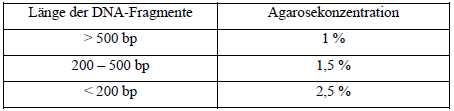
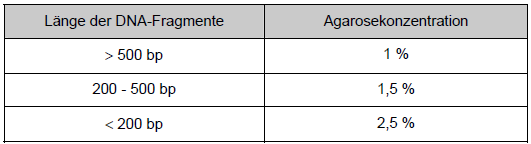
Die Agarosegelelektrophorese dient zur horizontalen Auftrennung von Doppelstrang- DNA nach ihrer Größe in einem elektrischen Feld, während bei einzelsträngigen Nukleinsäuren die Sekundärstruktur zusätzlich eine Rolle spielt. Durch Vergleich mit einem Molekulargewichts-Standard (Leiter) kann die Größe ermittelt und die Konzentration grob abgeschätzt werden. Zusätzlich ist eine präparative Gewinnung eines bestimmten DNA-Fragments durch Ausschneiden der entsprechenden Bande aus dem Gel nach erfolgter Auftrennung und anschließende Aufreinigung möglich. Sichtbar werden die Banden durch Interkalation des orange fluoreszierenden Farbstoffs Ethidiumbromid, der zur Gellösung und/oder zum Laufpuffer zugegeben wird. Verwendet werden Agarosekonzentrationen von 1 – 2,5 % (w/v) je nach Größe der aufzutrennenden Fragmente. Im Einzelnen werden folgende Konzentrationen verwendet:
Tab. 6: Übersicht über die Agarosekonzentration Die entsprechende Menge Agarose in 200 ml 1 x Laufpuffer wurde durch Kochen in einem Mikrowellenofen vollständig gelöst, μ3l [sic] Ethidiumbromid (10 mg/ml) zugegeben und gemischt. Zwei Gelkämme mit 20 Zähnen wurden eingesetzt. Nach Erstarren wurden die Kämme vorsichtig gezogen und das Geltablett in die mit 1 x Laufpuffer gefüllte Gelkammer gestellt. Die DNA-Probe wurde mit Auftragspuffer gemischt und aufgetragen. Zusätzlich wurden in einer am Rand gelegenen Spur 20 μl einer 100 bp-Leiter als Größenstandard aufgetragen. Der Lauf erfolgte bei 100 Volt für zwei Stunden. Das Gel wurde schließlich auf dem Tansilluminator [sic] betrachtet und photographiert. |
6.2.6 Agarosegelelektrophorese
Die Agarosegelelektrophorese dient zur horizontalen Auftrennung von Doppelstrang-DNA nach ihrer Größe in einem elektrischen Feld, während bei einzelsträngigen Nukleinsäuren die Sekundärstruktur zusätzlich eine Rolle spielt. Durch Vergleich mit einem Molekulargewichts-Standard (Leiter) kann die Größe ermittelt und die Konzentration grob abgeschätzt werden. Zusätzlich ist eine präparative Gewinnung eines bestimmten DNA-Fragments durch Ausschneiden der entsprechenden Bande aus dem Gel nach erfolgter Auftrennung und anschließende Aufreinigung möglich. Sichtbar werden die Banden durch Interkalation des orange fluoreszierenden Farbstoffs Ethidiumbromid, der zur Gellösung und/oder zum Laufpuffer zugegeben wird. Verwendet werden eine Agarosekonzentrationen von 1 − 2,5 % (w/v) je nach Größe der aufzutrennenden Fragmente. Im einzelnen werden folgende Konzentrationen verwendet:
Tabelle 2: Übersicht über die Agarosekonzentration Die entsprechende Menge Agarose (Sigma-Aldrich, Steinheim, Deutschland) in 200 ml 1 × Laufpuffer wurde durch Kochen in einem Mikrowellenofen vollständig gelöst, 3 μl Ethidiumbromid (10 mg/ml, Sigma) zugegeben und gemischt. Zwei Gelkämme mit 20 Zähnen wurden eingesetzt. Nach Erstarren wurden die Kämme vorsichtig gezogen und das Geltablett in die mit 1 × Laufpuffer gefüllte Gelkammer gestellt. Die DNA-Probe wurde mit Auftragspuffer [Seite 41] gemischt und aufgetragen. Zusätzlich wurden in einer am Rand gelegenen Spur 20 μl einer 100 bp-Leiter (Biozym, Hessisch Oldendorf) als Größenstandard aufgetragen. Der Lauf erfolgte bei 100 Volt für zwei Stunden. Das Gel wurde schließlich auf dem Transilluminator betrachtet und photographiert. |
Ein Verweis auf die Quelle fehlt. |
|
| [3.] Ves/Fragment 086 01 - Diskussion Zuletzt bearbeitet: 2014-07-01 14:52:56 Schumann | Fragment, Gesichtet, Mauermann 2004, SMWFragment, Schutzlevel sysop, Verschleierung, Ves |
|
|
|
| Untersuchte Arbeit: Seite: 86, Zeilen: 1 ff. (komplett) |
Quelle: Mauermann 2004 Seite(n): 41, 42, Zeilen: 41: 17 ff.; 42: 1 ff. |
|---|---|
| 3.2.7. Restriktions-Fragment-Längenpolymorphismus (RFLP)-Analyse
Restriktionsendonukleasen sind Enzyme, die von Bakterien synthetisiert werden und spezifisch DNA-Sequenzen erkennen und schneiden. Dabei wird zwischen Typ-I-, Typ-II- und Typ-III-Restriktionsendonukleasen unterschieden. Breite Anwendung finden vor allem die Typ-II-Restriktionsendonukleasen: Diese erkennen Palindrome und schneiden an genau definierten Positionen innerhalb der Erkennungssequenz. Palindrome sind in diesem Zusammenhang Sequenzen, welche mit ihrer Komplementärsequenz identisch sind. Liegt ein bestimmter Polymorphismus innerhalb einer Erkennungsstelle für ein bestimmtes Restriktionsenzym vor, so lässt er sich durch Restriktion (Verdau) eines PCR-Amplifikats nachweisen, welches die polymorphe Nukleotidposition beinhaltet. Ist keine komplette Erkennungsstelle vorhanden, kann durch eine in einzelnen Nukleotiden von der originalen Sequenz abweichende Primersequenz eine solche Erkennungsstelle in der PCR eingeführt werden. Voraussetzung dafür ist, dass in unmittelbarer Nachbarschaft der zu untersuchenden polymorphen Nukleotidposition eine zur Generierung einer Erkennungsstelle passende Sequenz vorliegt, die der Erkennungsstelle für ein bestimmtes Restriktionsenzym ausreichend ähnlich ist. Dabei muss die Nukleotidposition Teil der entsprechenden Erkennungsstelle sein (Erläuterung am Beispiel der 1007insC-Mutation des NOD2-Gens siehe unten). Der Ansatz für einen Restriktionsverdau beinhaltet einen bestimmten Puffer, das Restriktionsenzym sowie das zu verdauende PCR-Produkt. Es erfolgt eine Inkubation bei einer für jedes Enzym spezifischen Temperatur für mindestens zwei Stunden, meist jedoch über Nacht. Zur Visualisierung der entstandenen Restriktionsfragmente wird eine Agarosegelelektrophorese vorgenommen. 3.2.8. Genotypisierung des NOD2/CARD15-Gens in Bezug auf die 1007insC-Mutation Ziel der Untersuchung ist der Nachweis der 1007insC-Mutation im NOD2/CARD15- Gen. Diese Mutation besteht aus der Insertion eines C/G-Basenpaares an der Nukleotidposition 1007. Verwendet wurde ein PCR mit RFLP-Analyse. Entsprechend der umliegenden Sequenz wurde als Restriktionsenzym Mwol ausgewählt, dessen Erkennungssequenz ein unterbrochenes Palindrom der Sequenz GCNNNNN/NNGC ist, dabei bedeutet N jede beliebige Base, der Schnitt erfolgt zwischen den Nukleotiden sieben und acht [der Erkennungssequenz.] |
6.2.7 Restriktions-Fragment-Längenpolymorphismus (RFLP)-Analyse
Restriktionsendonukleasen sind Enzyme, die von Bakterien synthetisiert werden und spezifisch DNA-Sequenzen erkennen und schneiden. Dabei wird zwischen Typ-I-, Typ-II- und Typ-III-Restriktionsendonukleasen unterschieden. Breite Anwendung finden vor allem die Typ-II-Restriktionsendonukleasen. Diese erkennen Palindrome und schneiden an genau definierten Positionen innerhalb der Erkennungssequenz. Palindrome sind in diesem Zusammenhang Sequenzen, welche mit ihrer Komplementärsequenz identisch sind. Liegt ein bestimmter Polymorphismus innerhalb einer Erkennungsstelle für ein bestimmtes Restriktionsenzym, so lässt er sich durch Restriktion (Verdau) eines PCR-Amplifikats nachweisen, welches die polymorphe Nukleotidposition beinhaltet. Ist keine komplette Erkennungsstelle vorhanden, kann durch einen in einzelnen Nukleotiden von der originalen Sequenz abweichende Primersequenz eine solche Erkennungsstelle in der PCR eingeführt werden. Voraus- [Seite 42] setzung dafür ist, dass in unmittelbarer Nachbarschaft der zu untersuchenden polymorphen Nukleotidposition eine zur Generierung einer Erkennungsstelle passende Sequenz vorliegt, die der Erkennungsstelle für ein bestimmtes Restriktionsenzym ausreichend ähnlich ist. Dabei muss die Nukleotidposition Teil der entsprechenden Erkennungsstelle sein (Erläuterung am Beispiel der 3020insC-Mutation des NOD2-Gens s.u.). Der Ansatz für einen Restriktionsverdau beinhaltet einen bestimmten Puffer, das Restriktionsenzym sowie das zu verdauende PCR-Produkt. Es erfolgt eine Inkubation bei einer für jedes Enzym spezifischen Temperatur für mindestens zwei Stunden, meist jedoch über Nacht. Zur Visualisierung der entstandenen Restriktionsfragmente wird eine Agarosegelelektrophorese vorgenommen. 6.2.8 Genotypisierung des NOD2/CARD15-Gens in Bezug auf die 3020insC-Mutation Ziel der Untersuchung war der Nachweis der 3020insC-Mutation im NOD2/ CARD15-Gen. Diese Mutation besteht aus der Insertion eines C/G-Basenpaares an der Nukleotidposition 3020. Verwendet wurde eine PCR mit RFLP-Analyse. Entsprechend der umliegenden Sequenz wurde als Restriktionsenzym MwoI (New England Biolabs, Beverly, MD, USA) ausgewählt, dessen Erkennungssequenz ein unterbrochenes Palindrom der Sequenz GCNNNNN/NNGC ist, dabei bedeutet N jede beliebige Base, der Schnitt erfolgt zwischen den Nukleotiden sieben und acht der Erkennungssequenz. |
Ein Verweis auf die Quelle fehlt. |
|
| [4.] Ves/Fragment 087 01 - Diskussion Zuletzt bearbeitet: 2014-06-21 09:05:24 Hindemith | Fragment, Gesichtet, Mauermann 2004, SMWFragment, Schutzlevel sysop, Verschleierung, Ves |
|
|
|
| Untersuchte Arbeit: Seite: 87, Zeilen: 1 ff. (komplett) |
Quelle: Mauermann 2004 Seite(n): 42, 43, Zeilen: 42: 21 ff.; 43: 1 ff. |
|---|---|
| Das erste GC ist in der Originalsequenz vorhanden, während das zweite GC über den 3’-Primer in der PCR eingeführt wird. Dabei ist die Einführungsstelle so gewählt, dass die Erkennungssequenz nur dann entsteht, wenn die beschriebene Insertion vorliegt. Liegt im Falle des Wildtyps die Insertion hingegen nicht vor, entsteht anstatt der Erkennungssequenz GCN7GC die Sequenz GCN6GC, die keine Erkennungsstelle für das Enzym Mwol darstellt.
Folgende Primer wurden verwendet:
Die zwei unterstrichenen Basen im 3’-Primer entsprechen nicht der Originalsequenz, sie dienen der Einführung der Erkennungsstelle für Mwol.
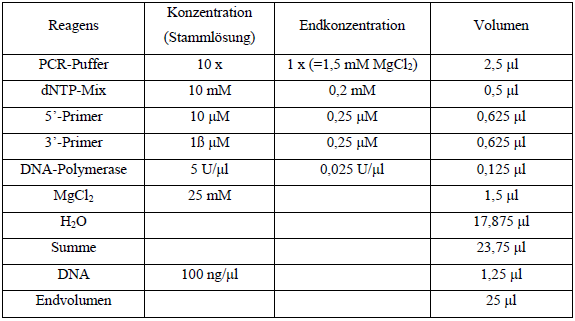
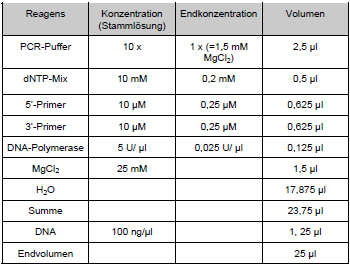
Tab. 7: PCR-Ansatz |
Das erste GC ist in der Originalsequenz vorhanden, während das zweite GC über den 3’-Primer in der PCR eingeführt wird. Dabei ist die Einführungsstelle so gewählt, dass die Erkennungssequenz nur dann entsteht, wenn die beschriebene Insertion vorliegt. Liegt im Falle des Wildtyps die Insertion hingegen nicht vor, entsteht statt der Erkennungssequenz GCN7GC die Sequenz GCN6GC, die keine Erkennungsstelle für das Enzym MwoI darstellt.
[Seite 43] Folgende Primer wurden verwendet (Hersteller: TIB MOLBIOL, Berlin, Deutschland):
Die zwei unterstrichenen Basen im 3’-Primer entsprechen nicht der Originalsequenz, sie dienen der Einführung der Erkennungsstelle für MwoI. [...]
Tabelle 3: PCR- Ansatz |
Ein Verweis auf die Quelle fehlt. |
|
| [5.] Ves/Fragment 088 01 - Diskussion Zuletzt bearbeitet: 2014-06-21 09:05:21 Hindemith | Fragment, Gesichtet, Mauermann 2004, SMWFragment, Schutzlevel sysop, Verschleierung, Ves |
|
|
|
| Untersuchte Arbeit: Seite: 88, Zeilen: 1 ff. (komplett) |
Quelle: Mauermann 2004 Seite(n): 44, 45, Zeilen: 44: 1 ff.; 45: 1 ff. |
|---|---|
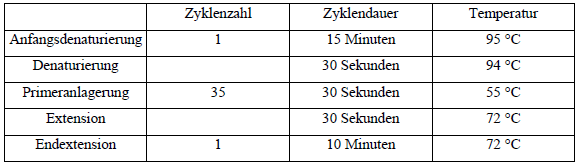
| Die PCR wurde in dem Thermocycler UNO Thermoblock, BIOMETRA®, Göttingen,
Deutschland unter folgenden Bedingungen durchgeführt:
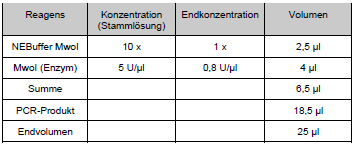
Tab. 8: PCR-Zyklen Um den Erfolg der PCR zu kontrollieren, wurden Stichproben der Amplifikate vor dem Restriktionsverdau mittels Agarosegelelektrophorese sichtbar gemacht. Die Gelkonzentration war 2,5 %. Es wurden jeweils 8 μl des PCR-Ansatzes gemischt mit 8 μl 1:1 verdünntem Auftragspuffer auf das Gel aufgetragen. Als Größenstandard wurden 2μ0l [sic] der 100 bp-Leiter verwendet. Das Gel wurde nach einer Stunde Laufzeit bei 100 V auf dem Transilluminator unter UV-Licht betrachtet und fotografiert. Die Restriktion erfolgte unter Verwendung des Enzyms Mwol. Für diese Reaktion wurde ein spezifischer Restriktionspuffer verwendet, die Inkubation erfolgte für zwei Stunden bis über Nacht bei 60 °C in einem Hybridisierungsofen. Der Restriktionsansatz setzte sich wie folgt zusammen:
Tab. 9: Zusammensetzung Restriktionsverdauung |
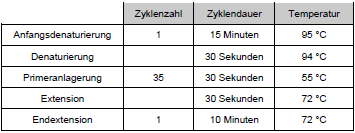
Die PCR wurde in dem Thermocycler UNO Thermoblock, BIOMETRA®, (Göttingen, Deutschland) unter folgenden Bedingungen:
Tabelle 4: PCR - Zyklen Um den Erfolg der PCR zu kontrollieren, wurden Stichproben der Amplifikate vor dem Restriktionsverdau mittels Agarosegelelektrophorese sichtbar gemacht. Die Gelkonzentration war 2,5 %. Es wurden jeweils 8 μl des PCR-Ansatzes gemischt mit 8 μl 1:1 verdünntem Auftragspuffer auf das Gel aufgetragen. Als Größenstandard wurden 20 μl der 100 bp-Leiter verwendet. Das Gel wurde nach einer Stunde Lauf bei 100 V auf dem Transilluminator unter UV-Licht betrachtet und fotografiert (s. Anhang Abb.18). Die Restriktion erfolgte unter Verwendung des Enzyms MwoI (New England Biolabs, Beverly, MD, USA). Für diese Reaktion wird ein spezifischer Restriktionspuffer verwendet, die Inkubation erfolgte für zwei Stunden bis über Nacht bei 60 °C in einem Hybridisierungsofen (Hybridizer HB-1D, Techne, Cambridge, UK). Der Restriktionsansatz setzte sich wie folgt zusammen: [Seite 45]
Tabelle 5: Zusammensetzung Restriktionsverdau |

Ein Verweis auf die Quelle fehlt. |
|
| [6.] Ves/Fragment 089 01 - Diskussion Zuletzt bearbeitet: 2014-06-21 09:05:18 Hindemith | Fragment, Gesichtet, Mauermann 2004, SMWFragment, Schutzlevel sysop, Verschleierung, Ves |
|
|
|
| Untersuchte Arbeit: Seite: 89, Zeilen: 1-8 |
Quelle: Mauermann 2004 Seite(n): 45, Zeilen: 1 ff. |
|---|---|
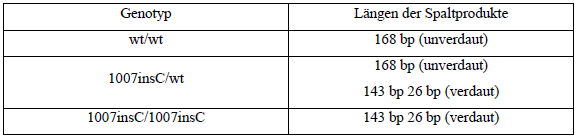
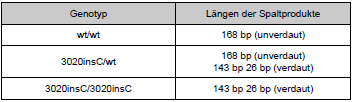
| Nach Verdau des PCR-Produkts mit Mwol entstanden Produkte folgender Längen (wt: Wildtyp):
Tab. 10: Produkte nach Restriktionsverdau Die Visualisierung der Restriktionsfragmente erfolgte mittels Agarosegelelektrophorese. Die Gelkonzentration war hierbei ebenfalls 2,5 %. Es wurden jeweils 10 μl des Restriktionsansatzes gemischt mit 8 μl 1:1 verüdnntem Auftragspuffer auf das Gel aufgetragen. Das Gel wurde nach zwei Stunden Laufzeit bei 100 V auf dem Transilluminator unter UV-Licht betrachtet und fotografiert. Als Größenstandard wurden die 100 bp-Leiter verwendet. |
Nach Verdau des PCR-Produkts mit MwoI entstehen Produkte folgender Längen (wt: Wildtyp):
Tabelle 6: Produkte nach Restriktionsverdau Die Visualisierung der Restriktionsfragmente erfolgte mittels Agarosegelelektrophorese. Die Gelkonzentration war hierbei ebenfalls 2,5 %. Es wurden jeweils 10 μl des Restriktionsansatzes gemischt mit 8 μl 1:1 verdünntem Auftragspuffer auf das Gel aufgetragen. Das Gel wurde nach zwei Stunden Lauf bei 100 V auf dem Transilluminator unter UV-Licht betrachtet und fotografiert. Als Größenstandard wurde die 100 bp-Leiter verwendet. |
Ein Verweis auf die Quelle fehlt. |
|