|
|
|
| Untersuchte Arbeit: Seite: 70, Zeilen: 1 ff. (complete page) |
Quelle: Anézo 2003 Seite(n): 156, 157, Zeilen: 156: 13ff; 157:1-9 |
|---|---|
| [At the] beginning of the simulation, the solute molecules are treated as point masses with zero Lenard-Jones parameters, so that they do not interact with the lipid environment. Step by step, the Lenard-Jones parameters and charges are rescaled, so that the solute molecules fully interact with the lipids at the endof [sic] the simulation. Lenard-Jones and electrostatic interactions from the initial state to the final state are computed using the following non bonded potential:
where λ = 0 corresponds to the initial state (i.e. no interaction between the solute and the membrane) and λ = 1, to the final state (i.e. full interaction). The parameter λ is incremented each time step, so that the solute molecules are progressively “grown” in size. This procedure allows one to incorporate the solute molecules without having first to create free volume, which would induce large perturbations in the bilayer structure. Note that the solute coordinates are “frozen” during the whole procedure to maintain the initial solute distribution throughout the bilayer. Three equilibrium MD simulations of 35 ns are carried out with each type of solute in the DPPC membrane. Simulation conditions are the same as those for pure DPPC system, i.e. ESFF, NPT ensemble with T = 323K and Pxx = Pyy= Pzz = 1 bar, choice of PME for the computation of electrostatics and PBC. All bond lengths are kept constant and time step was 2 fs. Equilibration time of 5 ns was required for all simulations, so that trajectories are analyzed over 30 ns simulation time. |
[page 156]
At the beginning of the simulation, the solute molecules are treated as point masses with zero LJ parameters and charges, so that they do not interact with the lipid environment. Step by step, the LJ parameters and charges are rescaled, so that the solute molecules fully interact with the lipids at the end of the simulation. LJ and electrostatic interactions from the initial state (state A) to the final state (state B) are computed in the following way (see Chapter 3, Section 3.1.3, page 56, for the notations):
where λ = 0 corresponds to the initial state (i.e. no interaction between the solute and the membrane) and λ = 1, to the final state (i.e. full interaction). The parameter λ is incremented each time step, so that the solute molecules are progressively “grown” in size. This procedure allows one to incorporate the solute molecules without having first to create free volume, which would induce large perturbations in the bilayer structure. Note that the solute coordinates are “frozen” during the whole procedure to maintain the initial solute distribution throughout the bilayer. [page 157] Three equilibrium MD simulations of 30 ns are carried out with each type of solute in the DPPC membrane. Simulation conditions are the same as those for the pure DPPC system (see Chapter 4, Section 4.2, page 78), i.e. NPT ensemble with T = 323 K and Pxx = Pyy = Pzz = 1 bar, choice of PME for the computation of electrostatics, and periodic boundary conditions. All bond lengths in the solute molecules are kept constant using the LINCS algorithm [109]. The time step is decreased from 5 to 2 fs, however, to ensure a more accurate description of solute dynamics. An equilibration time of 2 ns was required for the simulations with methylglucose and mannitol, and 5 ns with salicylic acid, so that trajectories are analyzed over 28 and 25 ns simulation time, respectively. |

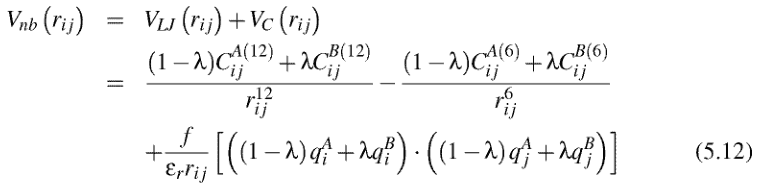
No source is given. |
|